Université de Liège - Centre de l'Oxygène, Recherche et Développement (CORD)
Université de Liège - Centre de l'Oxygène, Recherche et Développement (CORD) Université de Liège - Centre de l'Oxygène, Recherche et Développement (CORD)
L'oxygène et la vie: tome 1 - Initiation au métabolisme de l'oxygène
C Deby et G Deby-Dupont
1. Définition simple d'un métal de transition
Les métaux de transition ont été ainsi appelés parce qu'ils semblaient assumer la transition entre les éléments à caractères métalliques très prononcés et les non-métaux (anciennement métalloïdes). Dans le tableau de Mendeleïeff, ils constituent un pont entre les deux classes d'éléments.
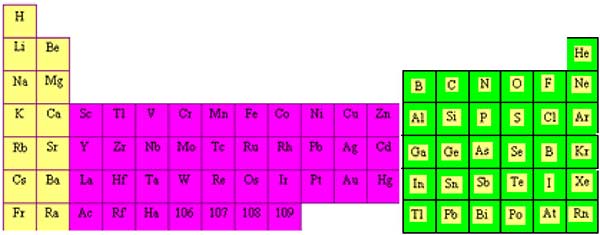
Fig. VIII-1 : La passerelle des métaux de transition (en violet)
La caractéristique principale des métaux de transition est de présenter des orbitales d incomplètement saturées en électrons. Les 5 orbitales d se remplissent progressivement par acquisition de 1 à 10 électrons, selon une des règles de Hund.

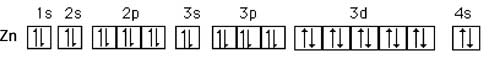
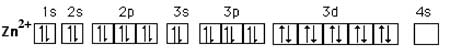
Fig. VIII-2 : Un exemple d'orbitale 3d complètement remplie : le zinc.

Fig. VIII-3 : Des orbitales 3d incomplètement remplies : le fer.
Les orbitales 3d du fer non ionisé sont incomplètement remplies : elles ne contiennent que 6 électrons (le maximum pour cette couche étant 10, comme le rappelle l'exemple précédent). Cette particularité explique le caractère paramagnétique intense du fer. Le nombre d'électrons célibataires atteint un maximum dans les cas de Fe3+ : 5.
2. Formation des complexes de coordination
Une autre caractéristique des métaux de transition est la facilité à former des complexes avec des molécules porteuses de paires d'électrons, les ligands. Ceux-ci s'unissent aux métaux de transition par un type de liaison particulière, dite de coordination (ou liaison dative), nettement plus faible que la liaison de covalence. La liaison de coordination est une mise en commun d'une ou plusieurs paires d'électrons, en provenance de la couche 2S du ligand, avec des orbitales vides du métal.
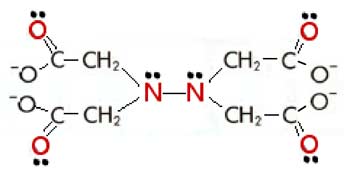
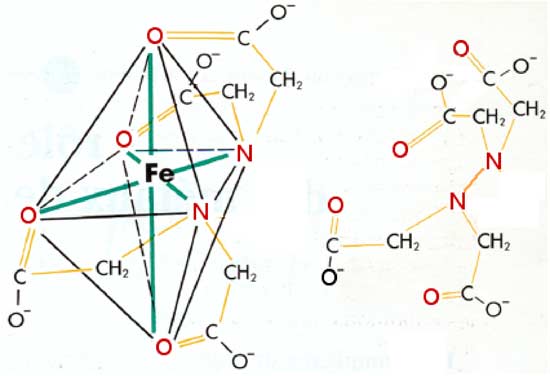
| Fig. VIII-6 : A gauche : le complexe EDTA-fer. Le fer possède 6 sites de coordination (traits verts), tous occupés ici par les atomes "loueurs" de paires d'électrons, colorés en rouge. Noter que l'étage d'oxydation du fer n'est pas indiqué, car il peut être 2 ou 3; pour être complexé, le métal doit évidemment être ionisé. A droite: la molécule d'EDTA est figurée "détachée" de l'image de gauche. |
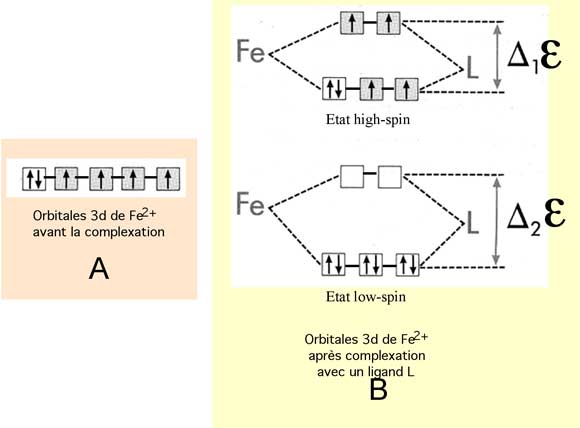
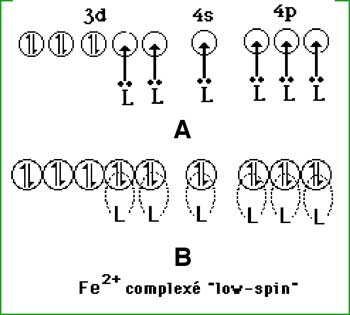
Fig. VIII-7 : Evolution de la multiplicité d'un métal selon la nature du ligand
(Revoir multiplicité au chapitre III, 5c ; explications dans le texte ci-dessous).
Avant la complexation, les orbitales d sont au même niveau énergétique (on dit qu'elles sont dégénérées) (fig. VIII-7 A). La complexation lève la dégénérescence et clive le niveau énergétique en 2 sous-niveaux (fig. VIII-7B). Généralement, 3 orbitales sont sur le sous-niveau le plus bas : c'est le cas de la fig.VIII-7B.
Deux cas sont possibles et sont représentés sur la fig. VIII-7 B:
a) la complexation a libéré une faible énergie ε ; les sous-niveaux sont séparés par une faible différence d'énergie Δ1ε et les électrons suivent la règle de Hund en se répartissant sur un maximum d'orbitales : c'est l'état high-spin. Pour le Fe2+ qui compte 6 électrons 3d, 4 électrons célibataires sont produits. Le fer est paramagnétique (ferromagnétisme).
b) ε est plus élevée; Δ2ε est plus grande que Δ1ε; les électrons restent sur le sous-niveau le plus bas : c'est l'état low-spin. Le Fe2+ rassemble tous ses électrons sur les 3 orbitales basses; tous les électrons sont appariés; le paramagnétisme fait place au diamagnétisme.
Dans le cas du fer ferrique Fe3+ qui ne compte plus que 5 électrons 3d, il n'y aura qu'un électron célibataire dans le complexe low-spin, avec un paramagnétisme plus faible.
L'énergie de clivage (ou de splitting) varie selon la nature du ligand : l'EDTA crée un champ faible et un état high-spin. L'anion cyanure fait l'inverse.
4. Interactions des métaux de transition avec O2L'oxygène se lie facilement aux métaux de transition. Les éléments de transition apparaissent comme des intermédiaires indispensables entre l'oxygène triplet et les singulets, par une possibilité de présence, dans leurs orbitales 3d, d'électrons célibataires.
a. Activité oxydante des complexes métalliques
La complexation conserve la plupart des propriétés initiales des partenaires, mais peut modifier leur capacité oxydante. Les liaisons de coordination sont plus lâches que les covalences, mais induisent chez les métaux de transition un remaniement électronique facilitant la donation d'électrons en provenance du métal vers l'oxygène. L'oxygène est lié transitoirement à un atome de métal de transition (en biologie, il s'agit principalement de Fe), cette liaison étant associée à un transfert d'électrons du métal vers O2. Ce phénomène exige la présence de molécules ou de sites moléculaires formant avec le métal un complexe de coordination actif.L'oxygène coordonné à un métal peut devenir très réactionnel selon le ligand.
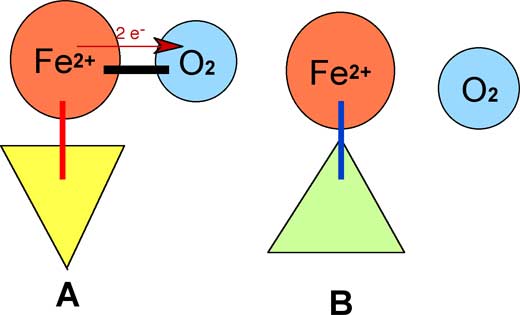
Fig. VIII-8 : A Complexe ternaire où un ligand (triangle jaune) rassemble un atome de fer ferreux et une molécule d'oxygène. Le ligand favorise le transfert (flèche rouge) de 2 électrons vers l'oxygène : complexe actif.
B Le ligand (triangle vert) ne favorise pas le transfert d'électrons : complexe inactif qui ne retient pas l'oxygène.Un exemple d'oxydation in vitro par le fer : la réaction d'Udenfriend
Une des nombreuses fonctions hépatiques est le catabolisme de substances toxiques et de nombreux médicaments, comme les barbituriques. Cette "détoxification" est assurée notamment par un processus d'hydroxylation. Avant la découverte du système du cytochrome P450, divers modèles chimiques furent proposés pouvant expliquer cette mystérieuse capacité du foie. Udenfriend et coll. découvrirent en 1954 que le système Fe(II)-EDTA/ascorbate/O2 peut hydroxyler divers composés, non seulement aromatiques (phénols par ex.), mais aussi aliphatiques saturés. Mais on se rendit promptement compte de la difficulté de diriger cette réaction et d'en donner un mécanisme exact. D'autres métaux de transition (comme le cuivre) peuvent remplacer le fer. D'autres réducteurs que l'ascorbate (comme les tétrahydroptéridines) peuvent être employés avec la même efficacité.
Bref, on constata que les métaux de transition peuvent participer à des systèmes d'oxydation en servant d'intermédiaires entre l'oxygène et le substrat oxydable, en présence d'un réducteur de potentiel redox bien déterminé.
b. Généralités sur les ferryls
Certains ligands permettent le transfert d'un électron (e-) du fer vers une molécule d'oxygène (après "fixation" de celle-ci par une des liaisons de coordination du fer):
L-Fe2+ + O2 ====> L-Fe2+......O2Les étapes suivantes sont représentées sur la fig. VIII-9. La liaison de coordination est du type "bent-on". Le transfert d'électron produit un radical intermédiaire, superoxo, devenant l'ion oxoferryle.
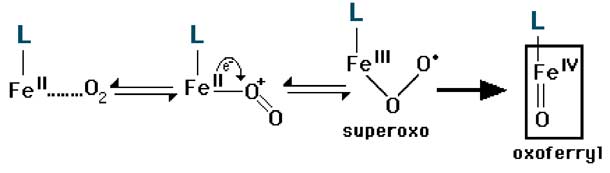
Fig. VIII-9 : Oxydation d'un atome de fer (FeII) par une molécule d'O2, par transfert d'électron.
L'ion oxoferryle est très réactionnel (voir fig. VIII-10 d).
Dans d'autres cas (dépendant toujours du ligand), l'atome FeII réduit l'oxygène par 2 électrons (fig. VIII-10 a) et forme l'ion perferryle, l'atome Fe montant au degré d'oxydation 4. Après la formation d'un complexe peroxo (fig. VIII-10 b), le pont -O-O- se casse et libère 2 ions oxoferryles, comme dans le premier cas (fig. VIII-10 c).

Fig. VIII-10 : Formation de l'ion oxoferryle très réactionnel.
L'ion oxoferryle est très réactionnel grâce à un atome d'oxygène uni par une liaison de coordination plus lâche qu'une covalence, à son état singulet et à sa haute densité électronique. Cette réactivité élevée se manifeste en milieu non-aqueux (intra-membranaire) comme en milieu aqueux; dans ce dernier cas, il pourrait se former, en réaction flash avec des protons H+, des radicaux •OH réagissant sur le lieu même de leur formation, cette probabilité de réaction étant en raison inverse du pH (d'après Sugimoto et Sawyer, 1985).
5. Rôle des métaux de transition en biologie
Les métaux de transition, notamment le fer, le cuivre, le manganèse, le cobalt et le molybdène, sont des catalyseurs des peroxydations lipidiques. Leur structure électronique leur permet d'être complexés par des ligands (ou chélateurs), au moyen de liaisons de coordination laissant libres leurs liaisons de valence. Les complexes métalliques ainsi formés peuvent activer l'oxygène par des mécanismes encore discutés en permettant la fixation de O2 sur des molécules organiques. Les réactions productrices de radicaux (fig. VIII-10) ne sont réalisables, de même que la réduction du métal, que si ce dernier est complexé par un ligand favorable. Certains ligands inhibent les phénomènes d'oxydation en rendant inutilisables du point de vue catalytique les métaux de transition (desferrioxamine, par exemple : voir chapitre XV).
Les hèmes, groupements prosthétiques d'un grand nombre d'enzymes, des cytochromes, de l'hémoglobine et de la myoglobine, sont constitués par un atome de fer complexé par une porphyrine (chapitre IX). Aux très faibles concentrations (ordre de la micromole), ils catalysent énergiquement la peroxydation des lipides (chapitre X). Des traces d'hémoglobine provoquent le rancissement du lard, c'est-à-dire la peroxydation des graisses non saturées de cet aliment.
Les interventions des métaux de transition en chimie biologique sont le thème du chapitre suivant.6. Bibliographie
Métaux de transition
Gerloch M, Constable EC. Transition metal chemistry. Editions VCH, Weinheim, New York, Tokyo, 2000, 211 p.Réaction d'Udenfriend
Hamilton GA. Chemical models and mechanisms for oxygenases. In : Molecular mechanisms of oxygen activation. O.Hayashi, ed., Academic Press, NY, London, 1974, pp.405-452.
Maskos Z, Rush JD, Koppenol WH. The hydroxylation of tryptophan. Archives of Biochemistry and Biophysics. 1992; 296: 14-20.Complexes fer-oxygène
Sugimoto H, Sawyer DT. Iron(II)-induced activation of hydroperoxides for the dehydrogenation and monooxygenation of organic substrates in acetonitrile. Journal of American Chemical Society 1985; 107: 5712-16.
Sheldon RA. History of oxygen activation. In : The activation and homogeneous catalytic oxidation. DHR. Barton et al., eds, Plenum Press, New York, 1993, pp. 9-30.