Université de Liège - Centre de l'Oxygène, Recherche et Développement (CORD)
Université de Liège - Centre de l'Oxygène, Recherche et Développement (CORD)
L'oxygène et la vie: tome 1 - Initiation au métabolisme de l'oxygène
C Deby et G Deby-Dupont
Université de Liège - Centre de l'Oxygène, Recherche et Développement (CORD)
L'oxygène et la vie: tome 1 - Initiation au métabolisme de l'oxygène
C Deby et G Deby-Dupont
1. Le fer dans les milieux biologiques
"Under physiological conditions, iron is only slightly soluble, tending to form precipitates with anions such as OH-". Graf et al., 1984
"Life, in any form, without iron, is in all likehood, impossible". Neilands, 1972a. Ubiquité du fer dans les tissus
Le fer est le métal de transition le plus répandu dans l'organisme des mammifères. Par exemple, le coeur contient 96 mg Fe/g de tissu calciné et le cerveau 63 mg (Perry et coll., 1962). La teneur en fer du cytosol se situe entre 10-5 et 1,3.10-4M, généralement autour de 5,5.10-5M (Fong et coll., 1976).
Le fer ionisé en systèmes aqueux
Les sels ferreux (Fe2+), dissous à pH neutre (tampon phosphates), s'oxydent rapidement en présence d'oxygène. Leur demi-vie est estimée à moins de 15 secondes dans le cas de solutions 10-3M dans un tampon 50 mM en phosphates, à pH 7,0 (Slivka et coll., 1986).
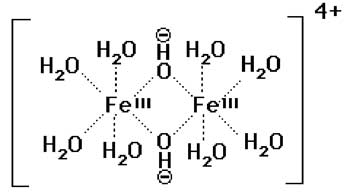
En systèmes aérobies, aux pH biologiques, le fer est au degré d'oxydation III, état stable dû à la présence de O2 (Aisen et Listowsky, 1980). Tout autre niveau d'oxydation nécessite une structure protectrice (protoporphyrine IX, par ex), maintenue par une fourniture d'énergie. En solution aqueuse, en l'absence de ligand, les ions Fe3+ deviennent très rapidement un complexe aqueux : Fe3+...8H2O. Ce complexe dimérise puis polymérise alors rapidement en plus grosses molécules, insolubles dans l'eau, qui forment la rouille (Rosen et Klebanoff, 1981).
Fig. IX-1 : Exemple de dimère intermédiaire de Fe3+...8(H2O).
Les atomes de fer sont unis par des ponts oxo.Nécessité d'un complexant
Le fer non complexé apparaît biologiquement inaccessible (Crapo, 1977). Les ligands naturels du fer, maintenant celui-ci à l'état soluble, sont très nombreux : albumine, acide ascorbique, … et surtout les porphyrines que nous étudions ci-dessous.
Existe-t-il du Fe3+ libre ?
La concentration du Fe3+libre, aux pH physiologiques, a été estimée aux environs de 10-17M (Spiro et Saltman, 1969).b. Oxydations par le fer complexé
1. La réaction de Fenton
A la fin du XIXe siècle, Fenton décrivait l'oxydation de l'acide tartrique par le système ion ferreux + H2O2. En 1931, Haber et Willstätter impliquèrent le radical hydroxyle dans la réaction décrite par Fenton. L'année suivante, Haber et Weiss proposaient l'équation suivante:
H2O2 + Fe2+ ===> Fe3+ + •OH + OH- (1)
La régénération de Fe2+ fut confiée à l'anion superoxyde qui réduit Fe3+.Pour les générations de chercheurs suivantes, l'équation (1) deviendrait la réaction de Fenton.
Depuis le début des années 1970, lorsqu'il fut avéré que H2O2 était produit par des systèmes enzymatiques in vivo, avec passage par la création d'anion superoxyde, de nombreux chercheurs, assurés que du fer ionisé est disponible dans le cytosol, réalisèrent des expériences in vitro, en systèmes chimiques purs, et montrèrent qu'effectivement, le système H2O2 + Fe2+ oxyde nombre de molécules d'importance biologique. Pour des centaines de chercheurs, •OH serait l'agent oxydant qui confère à la réaction de Fenton son efficacité oxydante. Des milliers de publications confessèrent ce credo : la réaction d'Haber et Weiss n'était qu'un bilan, mais la réaction de Fenton, une réalité biologique.
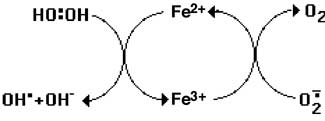
Fig. IX-2 : Le cycle de Fenton : H2O2 est réduit par le fer ferreux, libérant •OH.
Fe3+ est réduit en Fe2+ par l'anion superoxyde, et le cycle continue.
Remarquons qu'à cette époque, la complexation du fer par un ligand était superbement laissée de côté.
2. L'aliénation du fer par des corsets protéiques : transport et stockage du Fe
Potentiellement, les atomes ionisés de fer constituent une cause majeure des phénomènes de toxicité attribués à l'oxygène. Mais l'organisme et les cellules sont pourvues de protéines de transport et de stockage qui maintiennent ces ions au niveau d'oxydation III, les empêchant de catalyser des cycles d'oxydo-réduction dangereux.a) Transferrine : glycoprotéine plasmatique de 80 kDa. Principale responsable du transport du fer extracellulaire, elle lie 2 Fe3+ par molécule, auxquels sont associés un anion HCO3- par atome de fer. L'oxydation de Fe2+ est réalisée par des ferroxydases comme la céruloplasmine. Le complexe transferrine-Fe3+ entre dans les cellules de stockage par endocytose. Là, les atomes de Fe quittent la transferrine par abaissement du pH, pour être stockés par de grands édifices moléculaires constituant la ferritine. La molécule de transferrine quitte alors la cellule de stockage, prête à assumer un nouveau transport. Une même molécule de transferrine est apte à effectuer des milliers de voyages.
Il semble bien établi, après de nombreuses controverses, que, in vivo, le fer lié à la transferrine ne puisse pas participer aux réactions rédox.b) Lactoferrines : elles sont très voisines de la transferrine (glycoprotéines de 80 kDa, fixant 2 Fe3+ en présence de 2 HCO3-), mais ne participent pas au transport du Fe. Leur affinité pour le fer est bien plus élevée que celle de la transferrine : il faut descendre à pH 2 pour relâcher les atomes de Fe qu'elles complexent.
On distingue une lactoferrine des polynucléaires neutrophiles (voir chapitre XIII), des larmes, du fluide pancréatique et de la bile, et une lactoferrine du lait. Dans les polynucléaires, générateurs d'H2O2, le rôle de la lactoferrine serait d'éviter toute réaction de cet hydroperoxyde avec le fer. Dans le lait et les liquides muqueux, les lactoferrines ont essentiellement une fonction bactériostatique en privant les bactéries, contaminant éventuellement les surfaces muqueuses, du fer indispensable à leur croissance (ainsi agit par exemple la pénicilline). Les lactoferrines inhibent les réactions d'oxydation où est impliqué le fer.
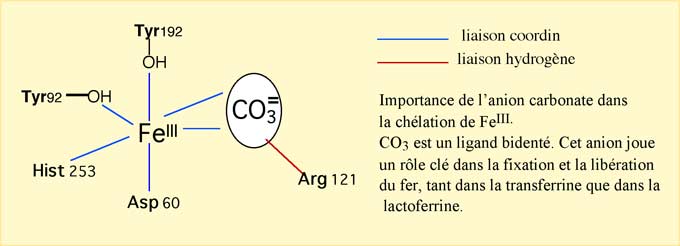
Fig. IX-3 : Un des deux sites de fixation du fer dans la lactoferrine humaine.
c) Le stockage par la ferritine : la ferritine est une protéine cytoplasmique extrêmement stable d'environ 450 kDa. Elle est constituée d'une coquille rassemblant 24 sous-unités semblables, éléments cylindroïdes creusés de canaux pénétrant dans la cavité centrale où est séquestré le Fe3+. Une molécule de ferritine peut stocker jusqu'à 4500 atomes de fer (fig. IX-4).
Rappelons qu'il existe une ferritine inductible dans les cellules endothéliales lors des phénomènes hémolytiques.
Destin du fer stocké dans les molécules de ferritine: il fut longtemps admis, d'après des expériences in vitro, que le fer stocké dans les molécules de ferritine pouvait être libéré par réduction en Fe2+, ce qui rendait impossible le séjour du métal dans la ferritine. Plusieurs agents réducteurs ont été proposés pour cette opération, particulièrement l'anion superoxyde et l'ascorbate.
On sait maintenant que des molécules de ferritine sont libérées dans le plasma; elles contiennent des ions Fe3+ moins nombreux que dans la ferritine cellulaire. Ces molécules entrent dans les cellules consommant le fer, notamment les lignées érythroblastiques de la moëlle osseuse responsables de la genèse des hématies. La ferritine est alors soumise à une protéolyse qui libère le fer au sein des érythroblastes. Une fois de plus, on constate la minutie avec laquelle le fer est séquestré dans ses passages dans l'organisme (fig. VIII-5).
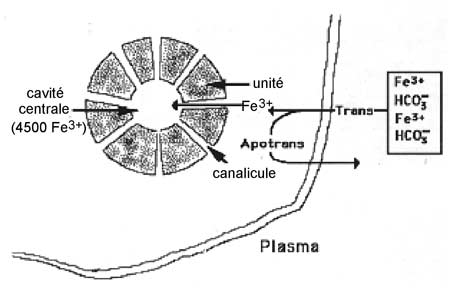
| Fig. IX-4 : Molécule de ferritine en coupe; les sous-unités ménagentes canalicules permettant le passage des ions Fe3+. Trans= transferrine |
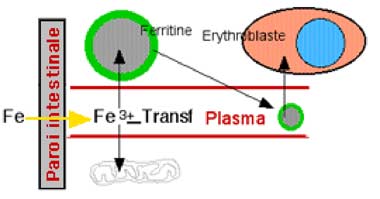
| Fig. IX-5 : Destinée du fer absorbé : fixation par la trans-ferrine (Transf) plasmatique, puis orientation vers les molécules de ferritine, vers les cellules-mères des globules rouge et vers les mitochondries qui synthétisent leurs cytochromes. |
d) Cas de saturation des sites de transport et de stockage : ce phénomène est tout à fait pathologique. Dans les conditions normales, il y a une importante désaturation de tous les types de sites. L'entrée du fer par absorption est strictement limitée au niveau de l'intestin, l'excès de fer alimentaire passant dans les fèces. Dans l'introduction à la pathologie de l'oxygène, on verra que des maladies telles que l'hémochromatose génétique ou acquise, ainsi que les phénomènes hémolytiques tels ceux qui se produisent de manière récurrente dans la thalassémie ou la drépanocytose (présence d'hémoglobine anormale fragilisant le globule rouge) aboutissent à une saturation des sites.
De même, on peut obtenir une saturation expérimentale par perfusion d'une solution d'un sel de fer complexé, par voie intra-veineuse ou intrapéritonéale.e) L'aliénation générale du Fe explique la concentration excessivement faible de fer libre calculée pour les tissus vivants : 10-17M.
3. La douteuse réalité de la réaction de Fenton in vivo
A présent, même dans un système simple, in vitro, on conteste l'existence de la réaction de Fenton mettant en jeu le système rédox Fe2+/Fe3+ et libérant des radicaux •OH en présence de peroxyde d'hydrogène.
1. Le fer libre est, normalement, complexé par des protéines qui l'empêchent de participer aux réactions d'oxydo-réduction.
2. Le fer ferreux ne peut subsister en solutions aqueuses : nous avons vu que ses 6 valences de coordination captent des ligands, tels que H2O, et que ce complexe se transforme rapidement en rouille insoluble et inactive.
3. Selon la nature du ligand, le fer sera actif ou non (voir diagrammes de splitting au chapitre VIII), c'est-à-dire porteur d'électrons célibataires.
4. Le fer actif serait à l'étage d'oxydation IV, à l'état d'espèces oxoferryles FeIV=O et perferryles FeIV-O2. Nous verrons au chapitre XIII que ces formes actives n'existent que dans des systèmes enzymatiques. Le fer n'agirait donc qu'incorporé dans ces enzymes.
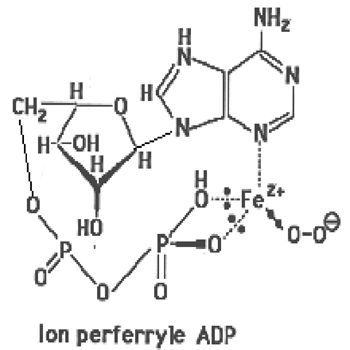
5. Toutefois, des auteurs ont affirmé que certains ligands naturels intracellulaires, comme l'acide adénosine diphosphorique, formé dans la chaîne respiratoire mitochondriale, pourraient complexer le fer et lui permettre de s'élever au niveau d'oxydation IV. Ainsi, le fer pourrait transférer des électrons vers l'oxygène sans nécessiter de supports protéiques (fig. IX-6).
Fig. IX-6 : Des complexes de ce type ont été mis en évidence dans les hématies.
4. Mécanisme proposé pour une formation d'ions ferryles non liée à des enzymes
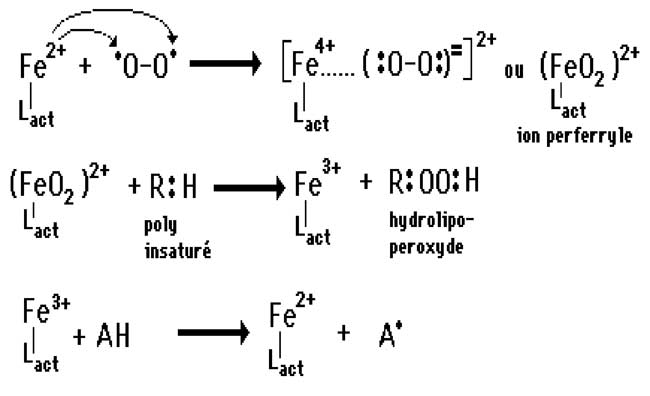
| Fig. IX-7 : Lact est un ligand actif tel que l'ADP, transférant 2 électrons de Fe2+ à l'oxygène avec lequel il forme un ion perferryle capable d'oxyder des molécules telles que les acides gras insaturés. Après cette oxydation, le fer est devenu ferrique et doit être réduit en Fe2+ par un réducteur AH (acide ascorbique, par ex.). |
Il est avéré que le fer participe à la plupart des réactions d'oxydation in vivo : c'est indéniable, mais c'est étroitement lié à des structures organiques soumises à de sévères processus de régulation que le fer agit. La plupart de ces processus engageant le fer nécessitent une structure hémoprotéique généralement enzymatique.
5. Structures hémoprotéiques et mécanismes d'oxydation
a. Structure de base du système actif : la porphyrine et l'hème qui est la porphyrine complexant un atome de fer ionisé.
La synthèse des hèmes se déroule dans les mitochondries.
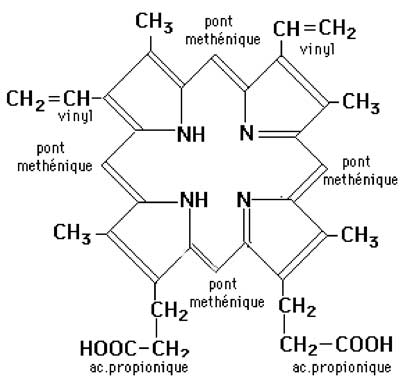
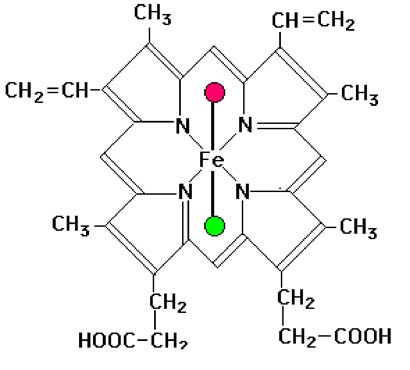
| Fig. IX-8 : La porphyrine IX (à gauche), commune à tous les mammifères, et son association à un atome de fer pour former un hème (à droite) dont nous voyons ici le modèle général. Le fer est hexacoordonné. Quatre liaisons le fixent aux 4 N des tétrapyrroles. Une des deux liaisons de coordination restantes l'arrime à un atome (N, S...) d'un résidu aminoacide de la protéine associée (vert) (résidu proximal), tandis que l'autre reste libre pour une coordination avec un ligand externe (rouge) qui est le résidu distal. |
Fig. IX-9 : Le macrocycle hémique : les atomes périphériques de l'hème forment un cercle constitué d'une alternance de simples et de doubles liaisons : - CH=CH-CH=CH- ..... Les doubles liaisons alternantes sont dites conjuguées et constituent un parcours de délocalisation d'un électron célibataire (résonance). L'enlèvement d'un atome d'hydrogène sur le macrocycle produit une structure radicalaire de vie suffisamment longue pour intervenir dans diverses réactions (voir chapitre IV).
b. Liaison de la protéine à l'hème : l'aminoacide de liaison détermine la nature de l'activité de l'hème. Deux types de structure sont remarquables pour les enzymes hémiques :
a) liaison proximale par un thiol SH,
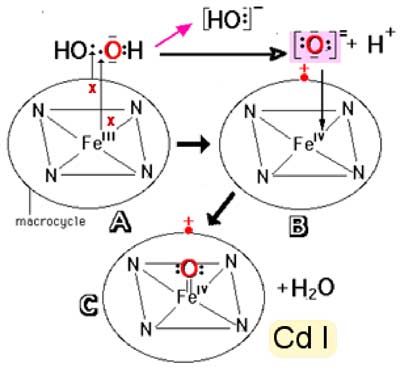
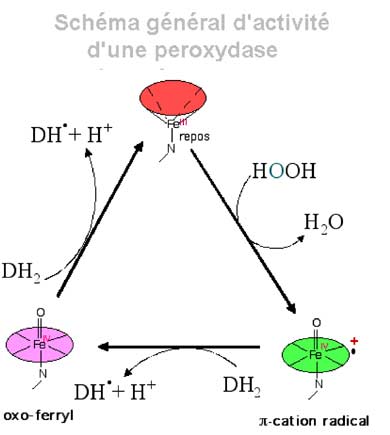
b) liaison proximale par une histidine.c. Cycle général d'oxydation : toutes les structures hémiques oxydantes passent par le cycle suivant, au cours duquel apparaît une structure radicalaire très active, le pi-cation radical.
Schéma des réactions : A-->B H2O2 + 2 e- -----> (OH)- + (:O:)= + H+
B--> C (:O:)= se lie par une double liaison au FeIV
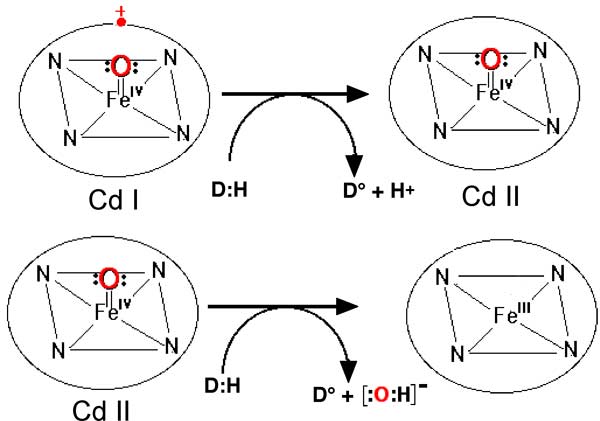
Fig. IX-11 : Retour à l'état fondamental et création de deux radicaux libres D• (Cd I: compound I)
| Fig. IX-12 : Cycle d'une peroxydase. Au cours de ce cycle, il y a formation de 2 radicaux libres DH•. a) Oxydation du fer hémique par H2O2 : L -FeIII + HOOH ----> H2O + L•+-FeIV=O L•+ est le macrocycle devenu radicalaire en perdant un électron, gagnant ainsi une charge positive (radical π-cation). b) Retour à FeIII: L•+-FeIV=O + D:H2 ----> L-FeIV=O + DH• + H+. L-FeIV=O + D:H2 ----> L-FeIII + DH• + OH- . OH- + H+ ----> H2O |
La fig. IX-12 représente un mécanisme général pour diverses enzymes oxydantes hémiques qui sont décrites dans le chapitre XIII.
6. Un cas particulier : l'autoxydation de l'hémoglobine
L'hémoglobine, transporteuse d'oxygène, peut dévier de cette fonction physiologique capitale par oxydation de son fer ferreux par l'oxygène transporté. Ce phénomène, découvert par Misra et Fridovich en 1972, est résumé dans l'équation suivante :
Hb-Fe2+....O2 ===> MetHb-Fe3+ + O2•
L'hémoglobine est une hémoprotéine tétramérique dont les atomes de fer sont maintenus, grâce à l'environnement protéique, à l'état d'oxydation minimale 2 (FeII). La liaison distale est réservée à la coordination d'une molécule d'oxygène. Dans certaines circonstances anormales (hémolyse par exemple), l'hémoglobine peut s'oxyder en méthémoglobine: les atomes de fer perdent un électron et deviennent FeIII. Dans ce cas, cette molécule bienfaisante devient un puissant catalyseur d'oxydation, fonctionnant comme une peroxydase (voir le chapitre X).
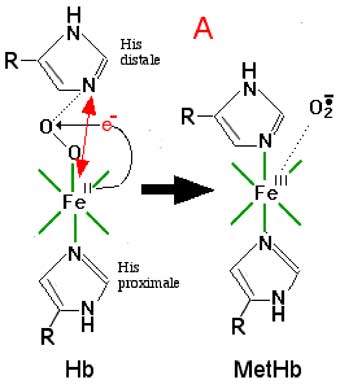
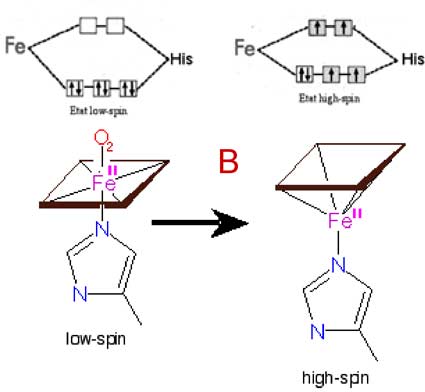
Fig. IX-13 : A : Un monomère d'hémoglobine Hb transporte une molécule d'oxygène, fixée par une des 6 liaisons de coordination. Seules les coordinations Fe-N du macrocycle sont représentées. Le complexe Fe2+-O2 est fixé par deux histidines de la globine, la proximale, coordonnant l'atome de Fe, et la distale, coordonnant l'oxygène transporté. Divers mécanismes ont été proposés pour expliquer comment, dans certaines conditions, Fe2+ peut céder un électron à O2 qui devient l'anion superoxyde. Une liaison de coordination s'établit alors entre l'histidine distale et le Fe3+ : Hb est devenue metHb, la methémoglobine.
B : Une des théories attribue l'autoxydation au changement de conformation électronique se produisant lors du départ de l'oxygène livré aux tissus. Le complexe Hb-O2 est diamagnétique, tous ses électrons étant appariés (low-spin). Après la perte d'O2, Hb devient paramagnétique et 4 de ses électrons sont alors célibataires. En même temps, le complexe porphyrine -Fe2+ passe de l'état plan à la configuration pyramidale. On admet que c'est au moment de la venue d'une nouvelle molécule d'oxygène que l'échange d'électron peut se produire. Mais des conditions supplémentaires sont requises dont l'exposition dépasse le cadre de cette initiation.La continuation de l'autoxydation par H2O2 découle de la dismutation de l'anion superoxyde en ce composé par qui les molécules d'oxyhémoglobine sont attaquées à leur tour (Everse et coll., 1994). La metHb formée fonctionne comme une peroxydase, selon le schéma de la fig. IX-12.
7. Les lipoxygénases
Ces enzymes ne sont pas hémiques; leur structure est brièvement décrite dans le chapitre XIII. Elles peroxydent les lipides non saturés.Mécanismes d'activité

2. Complexes du cuivre
Rôle du cuivre dans le métabolisme de l’oxygène
a. rôle dans la peroxydation lipidique
In vitro, dans la réaction d'Udenfriend, le fer peut être remplacé par le cuivre (voir chapitre X, sur la figure X-4 où le fer qui peut être remplacé par le cuivre).b. rôle anti-oxygène : les ferroxydases, molécules cuivriques
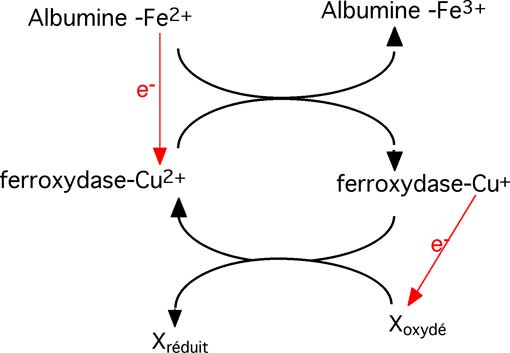
Ce sont des protéines plasmatiques qui oxydent le Fe2+, lui permettant d'être transporté par la transferrine.
Fig. IX-14 : Schéma général du fonctionnement d'une ferroxydase : le Fe2+ lié à l'albumine est oxydé par un atome de Cu2+ lié à une ferroxydase. Pour continuer son activité, celle-ci doit être soumise à un mécanisme de réoxydation où une molécule Xoxydé accepte l'électron du Cu+. 1) Céruloplasmine (ferroxydase I)
C’est une α2glycoprotéine bleue qui contient la plus grande partie du cuivre de l’organisme. Sa molécule contient plusieurs atomes de cuivre dont certains ne sont pas échangeables et d’autres disponibles pour l’échange. Chez l’homme, elle pèse 132 kDa et contient de 5 à 7 atomes de cuivre par molécule. La sécrétion de la céruloplasmine (Clp) par le foie est régulée par la glycosylation. La Clp augmente fortement dans l’inflammation, par stimulation de sa synthèse par les cytokines (IL1 et TNFα notamment). Les stéroïdes augmentent aussi la synthèse de la Clp par les hépatocytes.
Fonction: on lui a attribué différentes fonctions parfois opposées. Se basant sur des expériences in vitro, on a cru qu’elle favorisait les peroxydations. Aujourd’hui, in vivo, on lui attribue deux fonctions :
1. transport du cuivre (voir paragraphe suivant)
2. action anti-oxygène par sa capacité de jouer le rôle d’une ferroxydase.
« In vivo, ceruloplasmin would seem to be an all-purpose neutralizer of reactive oxygen species » (Linder et Goode, 1991, p. 93)
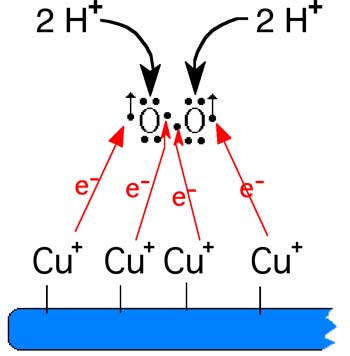
Dans le cas de la céruloplasmine, l'agent oxydant Xox est l'oxygène. Dès le début des recherches, on a constaté la nécessité, pour l'activité ferroxydasique de la céruloplasmine, de la présence d'oxygène. Comme pour la cytochrome oxydase, elle réduit O2 instantanément en H2O grâce à la présence de 4 atomes de cuivre voisins sur sa molécule :4 Cu+ + O2 + 4 H+===> 4 Cu++ + 2 H2O
Fig. IX-15 : Mécanisme d'oxydation du cuivre de la céruloplasmine.
La réduction de O2 est instantanée, sans stades activés intermédiaires.2) La ferroxydase II
Dans le sérum humain normal, elle représente 5% et moins de l’activité ferroxydasique totale. Elle comprend deux grandes sous-unités de 320 kDa et 220 kDa respectivement. La plus petite sous-unité contient un atome de cuivre par molécule, suffisant pour son activité ferroxydasique. Elle nécessite la présence d'oxygène, mais le mécanisme de transfert d'électrons n'est pas clairement expliqué.c. Transport plasmatique du cuivre
Le cuivre est absorbé généralement sous forme de carbonate de cuivre (CuCO3) et aussi, sous forme soluble dans l’eau, de sulfate de cuivre, de nitrate et de chlorure. L’absorption se fait dans le duodénum et le jéjunum. Elle semble moins réglementée que dans le cas du fer : on atteint donc plus facilement des intoxications que dans le cas du fer. Le cuivre absorbé apparaît d’abord dans le plasma sous forme d’ion cuprique (Cu2+) faiblement lié à l’albumine. Différentes molécules participent au transport du cuivre plasmatique :
- des protéines : 4 espèces
- céruloplasmine ou ferroxydase I
- albumine
- ferroxydase II
- transcupréine
- des oligopeptides et acides aminés
- rôle ambivalent joué par l'α2macroglobuline, transporteuse de zinc et de cuivre.
La principale destinée du cuivre absorbé est l’incorporation dans la céruloplasmine synthétisée dans le foie et aussi dans le réticulum endoplasmique de nombreux types de cellules. Le cuivre est excrété par le système biliaire : le taux normal sanguin est compris entre 50 et 150 µg/dL.
La transcupréine
C’est une protéine qui semble avoir une haute affinité pour le Cu2+ nouvellement absorbé. Sa masse moléculaire est 270 kDa.
Le cadre restreint de cette initiation ne nous permet pas d’envisager le rôle des oligopeptides et de l'α2macroglobuline.La superoxyde dismutase (SOD) à cuivre et à zinc
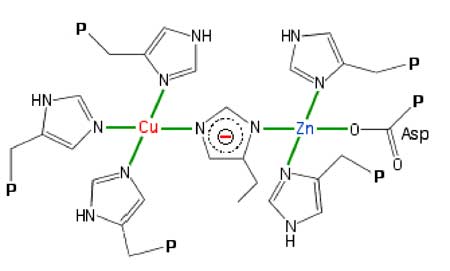
La SOD a une masse moléculaire de 32 kDa et est une protéine bleue à deux sous-unités. Au chapitre VII, nous avons défini le phénomène de dismutation qui est la fonction des SODs. Le site actif de la SOD est un cuivre complexé par 4 histidines dont l’une complexe également un atome de Zn. Cette histidine constitue un pont entre les deux atomes métalliques (fig. IX-16).
Fig. IX-16 : Site actif de la SOD Cu-Zn (simplifié d'après Potter et Valentine, 2003). Un atome de Cu est réuni par liaisons de coordination (vert) à un atome de Zn. Ce lien est le cycle imidazole d'une histidine déprotonée, dans lequel est délocalisé un électron (charge - ). Trois histidines achèvent de coordonner le Cu, tandis que deux autres, assistées d'une asparagine, complètent la coordination de Zn.
P : chaîne protéique
Cette curieuse structure a donné lieu à bien des controverses. Le zinc participe-t-il activement aux phénomènes rédox ? Est-il exact que le cycle imidazole qui sert de pont entre les deux métaux subisse des alternatives de protonation et de déprotonation ? Nous dépassons manifestement le cadre de cette initiation. Ce qui est certain, c'est l'existence d'un phénomène redox alternatif (fig. IX-17)
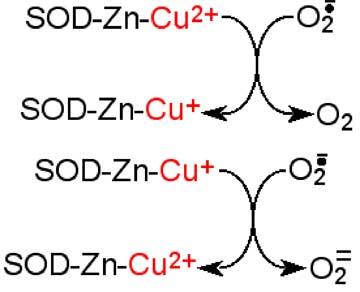
Fig. IX-17 : Mécanisme d'oxydoréductions alternatives, au sein de la SOD à Cu et Zn
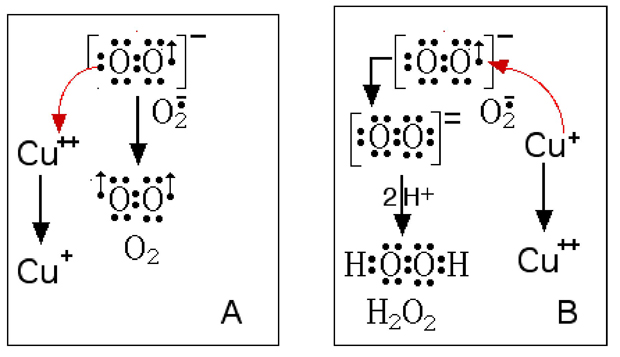
Fig. IX-18 : Transports d'électron au cours de l'activité de la SOD Cu-Zn,
aboutissant à la formation d'H2O2.
Sur la figure IX-18 sont représentées les deux réactions principales qui assument la fonction de dismutation. Au cours de la réaction A, le Cu(II) est réduit par l’anion superoxyde qui devient de l’oxygène ; dans la réaction B, le cuivre réduit cède un électron à une nouvelle molécule d’anion superoxyde qui, en présence de deux protons, génèrera H2O2.
Le rythme d'alternance des réactions est élevé et permet d’accélérer de plus de 1000 x la dismutation de l’anion superoxyde.
La fonction exacte du zinc n'est pas encore bien définie, car l'enlèvement de ce métal modifie peu l'activité de l'enzyme. D'autres SODs n'ont qu'un seul métal : celle des mitochondries ne possède qu'un atome de manganèse; beaucoup de SODs bactériennes fonctionnent avec un atome de fer.
3. Bibliographie
Propriétés et ubiquité du fer ionisé
Graf E, Mahoney JR, Bryant RG, Eaton JW. Iron-catalyzed hydroxyl radical formation. Stringent requirement for free iron coordination site. Journal of Biological Chemistry. 1984; 259: 3620-4.
Neilands JB. Structure & .Bonding (Berlin). 1972; 2: 145-170.
Perry HM Jr, Tipton IH, Schroeder HA, Cook MJ. Variability in the metal content of human organs. Journal of Laboratory and Clinical Medicine. 1962; 60: 245-53.
Fong KL, McCay PB, Poyer JL. Evidence for superoxide-dependent reduction of Fe3+ and its role in enzyme-generated hydroxyl radical formation. Chemical Biological Interactions, 1976; 15: 77-89.
Slivka A, Kang J, Cohen G. Hydroxyl radicals and the toxicity of oral iron. Biochemical Pharmacology. 1986; 35: 553-6
Aisen P, Listowsky I. Iron transport and storage proteins. Annual Review of Biochemistry. 1980; 49: 357-93.
Rosen H, Klebanoff SJ. Role of iron and ethylenediaminetetraacetic acid in the bactericidal activity of a superoxide anion-generating system. Archives of Biochemistry and Biophysics. 1981; 208: 512-9.
Crapo JD. The role of the superoxide dismutase in pulmonary oxygen toxicity. In : In : Superoxide and superoxide dismutases, Michelson AM, Mc Cord JM, Fridovich I, eds, Acad Press 1977, pp. 231-239.
Spiro TG, Saltman P. Structure & Bonding. Berlin. 1969; 6: 116-156.
Koppenol WH. The Haber-Weiss cycle--70 years later. Redox Report. 2001; 6: 229-34. Review.
Wardman P, Candeias LP. Fenton chemistry: an introduction. Radiation Research. 1996; 145: 523-31. Review.Cycle d'oxydation hémique
Meunier B, Bernadou J. Active iron-oxo and iron-peroxo species in cytochromes P450 and peroxidases; oxo-hydroxo tautomerism with water-soluble metalloporphyrins. In : Metal-oxo and metal-peroxo species in catalytic oxidations. Meunier B, ed, Springer, Berlin, 2000, pp.1-35.
Sheldon RA. History of oxygen activation. In : The activation and homogeneous catalytic oxidation. Barton DHR et al. eds, Plenum Press,` New York, 1993, pp. 9-30.
Fujii H. Electronic structure and reactivity of high-valent oxo iron porphyrins. Coordination Chemistry, 2002; 226: 51-60.Autoxydation de l'hémoglobine
Misra HP, Fridovich I. The generation of superoxide radical during the autoxidation of hemoglobin. Journal of Biological Chemistry. 1972; 247: 6960-2.
Everse J, Johnson MC, Marini MA. Peroxidative activities of hemoglobin and hemoglobin derivatives. Methods in Enzymology. 1944; 231: 547-561.Transport et stockage du fer
Testa U. Proteins of iron metabolism. CRC Press, Boca Raton London New York, 2002, 559 p.
Baker EN, Baker HM, Kidd KD. Lactoferrin and transferrin: functional variations on a common structural framework. Biochemical Cell Biology. 2002; 80: 27-34.
Eaton SS, Dubach J, Eaton GR, Thurman G, Ambruso DR. Electron spin echo envelope modulation evidence for carbonate binding to iron (III) and copper (II) transferrin and lactoferrin. Journal of Biological Chemistry 1990; 265: 7138-41.Biochimie du cuivre
Malmström BG, Leckner J. The chemical biology of copper. Current Opinions in Chemical Biology. 1998; 2: 286-292.
McGuirl MA, Dooley DM. Copper-containing oxidases. Current Opinions in Chemical Biology. 1999; 3: 138-144.
Linder MC, Goode CA. Biochemistry of copper. Plenum Press, New York London, 1991, 525 p.
Potter SZ, Valentine JS. The perplexing role of copper-zinc superoxide dismutase in amyotrophic lateral sclerosis. Journal of Biological and Inorganic Chemistry. 2003; 8: 373–380.