 Université de Liège - Centre de l'Oxygène, Recherche et Développement (CORD)
Université de Liège - Centre de l'Oxygène, Recherche et Développement (CORD) Université de Liège - Centre de l'Oxygène, Recherche et Développement (CORD)
L'Oxygène et la Vie: Tome II - L'Oxygène en Pathologie des Mammifères
Mitochondries et métabolisme de l'oxygène
Seconde partie : Troubles de l'oxygénation et mitochondries
Carol Deby
Chapitre V: Synopsis du rôle des ROS dans les affections cardiaques
| Note: pour la facilité de la lecture, 1. chaque référence dans le texte comporte un lien vers les pages de bibliographie 2. les abréviations et les formules chimiques sont reprises dans les pages du glossaire ; elles sont également identifiées directement dans le texte (apparition en arrière plan lors du pointage de la souris) |
C’est en 1997 que la revue Journal of Thrombosis and Thrombolysis consacra un numéro spécial aux phénomènes de reperfusion pouvant entraîner la mort.
Does « Lethal Reperfusion Injury» exist ? s’interrogeaient Venturini et Schaer, dans un article d’ouverture (Venturini et Schaer, 1997).
Le concept de lésion de reperfusion est-il une question importante dans une perspective clinique, se demandaient ces auteurs qui reconnaissaient que le « stunning » (systole continue réversible postischémique) pouvait conduire à l’issue fatale. Les mêmes reconnaissaient le rôle prépondérant des ROS, plus importantes que les polynucléaires neutrophiles trouvés en abondance dans le myocarde lors des autopsies.
Mais la controverse continuait, soit en affirmant que les causes de mort étaient présentes avant la reperfusion (Becker, 1997), soit en considérant que les dommages dus au phénomène de reperfusion était sans intérêt clinique. De manière générale, il était admis que deux mécanismes pathologiques pouvaient intervenir : l’apparition de radicaux libres durant les premières minutes du « reflow » et un trouble grave de l’homéostasie du calcium (Ferrari et Hearse, 1997). Dès cette époque, on envisageait l’application en clinique humaine du préconditionnement, sans toutefois être bien fixé sur les méthodes à utiliser pour le réaliser (Kukreja et Janin, 1997).
Mais personne, dans ce tour d’horizon de 1997, ne semblait penser aux mitochondries.
En 2001, des chercheurs, reconsidérant le problème du no-reflow, ne se préoccupaient toujours pas de ces organelles (Eeckhout et Kern, 2001; Iwakura et al., 2001) qui étaient cependant considérées par certains biochimistes comme les sources principales de ROS.
A. Les retombées de l’usage de la cyclosporine
Utilisée depuis largement 1989 comme immunosuppresseur, la cyclosporine A fut promptement reconnue comme protectrice des mitochondries lors des essais d’ischémie-reperfusion (Crompton et al., 1988; Broekmeier et al., 1989; Halestrap et Davidson, 1990; Griffiths et Halestrap, 1993).
Déjà à cette époque, la notion de pore mettant en communication l’intérieur de la mitochondrie et le cytosol se dégageait et son rôle dans l’apoptose devenait évident. Dès 1995, Griffiths et Halestrap démontraient qu’au cours de l’ischémie-reperfusion, c’est durant la phase de reflow que s’ouvre ce pore non spécifique, phénomène provoqué, semblait-il, par un excès de calcium matriciel ; les auteurs montraient que le rôle bénéfique de la cyclosporine s’exerçait par inhibition du pore nommé aujourd’hui mPTP, et plus précisément par liaison avec la cytophiline matricielle, impliquée dans l’ouverture du pore (Griffiths et Halestrap, 1995). En 1998, Griffiths et al. expliquèrent que les désordres graves entraînés par la reperfusion et améliorés par la cyclosporine étaient d’abord initiés par la rupture de l’homéostasie calcique, puis par la mise en panne de la production d’ATP, nécessaire, en autres, au fonctionnement des pompes calciques (Griffiths et al., 1998). Le rôle de la cytophiline D dans l’ouverture du mPTP des mitochondries cardiaques est bien étudié par l’équipe de Crompton (Johnson et al., 1999).
B. Les Francs-Tireurs
En 1982, Tanaka et al. signalaient les propriétés favorables du coenzyme Q10 (CoQ10) sur la récupération du myocarde après remplacement des valvules, chez l’homme. En 1996, Taggart et al. confirmaient les effets bénéfiques du CoQ10 sur les suites opératoires, mais ces chercheurs ne voyaient dans cet « antioxydant » qu’une sorte de vitamine liposoluble répandue dans les graisses animales.
En 2005, les résultats cliniques obtenus antérieurement avec le CoQ10 recevaient une nouvelle confirmation, mais les auteurs rappelaient la participation de cette molécule dans la chaîne de transport d’électrons mitochondriale et démontraient que le CoQ10 rentrait bien dans les mitochondries ; ces chercheurs signalaient en outre que le taux de CoQ10 diminue avec l’athérosclérose (Rosenfeld et al., 2005).
Rappelons que, depuis 1990, on sait que les statines font décroître le taux du CoQ10 chez le rat (Willis et al., 1990), par une inhibition de sa biosynthèse (Rundek et al., 2004). Marcoff et al. (2007) avertissent que les fonctions mitochondriales peuvent être perturbées au cours d’un traitement par statines et qu’il n’est pas certain que des suppléments alimentaires en CoQ10 puissent remédier à l’inhibition de la synthèse de cet intermédiaire de la chaîne ETC.
Il a donc fallu presque 20 ans pour que les cliniciens s’aperçoivent qu’un agent protecteur, agissant soi-disant comme antioxydant, jouait un tout autre rôle et que ses effets protecteurs agissaient dans les mitochondries.
2. Le point actuel
Les sujets de controverse s’amenuisent, devenant des points de détail. Quant aux grands mécanismes invoqués, ils ne sont plus contestés.
Le démarrage des lésions se produit lors de la reperfusion (Piper et al., 2004). Le départ a lieu dans les mitochondries, par altération de la phosphorylation oxydative faisant chuter la production d’ATP. Il se produit une élévation du calcium et l’ouverture du mégapore (mPTP), laissant échapper le cytochrome c, avec peroxydation des cardiolipines. La présence du cytochrome c dans le cytosol amorce les cascades d’événements biochimiques conduisant à l’apoptose.
Préoccupation actuelle majeure : inhiber le déclenchement de l’apotose cellulaire par lésion mitochondriale.
La position stratégique bien admise : le mPTP
Essayer d’empêcher son ouverture est le sujet principal dans ce domaine de la protection cardiaque (Halestrap et al., 2007; Halestrap, 2009).
A. Rôle du canal K+ATP
Ce sont des études récentes sur le mécanisme du préconditionnement ischémique qui ont conduit à l’émergence du rôle joué par l’activation de kinases par des ROS sortant par ce canal (Hausenloy et Yellon, 2004).
La voie du RISK (Reperfusion Injury Salvage Kinase)
Les kinases sont des enzymes impliquées dans la transmission des signaux au sein des cellules, formant des cascades par phosphorylation : une première kinase activant une seconde par phosphorylation ; cette dernière peut alors phosphoryler une troisième, jusqu’à la rencontre d’une molécule réceptrice finale.
Les kinases cytosoliques Akt (désignées aussi sous le sigle PKB : Protéine Kinase B) ainsi que les kinases ERK 1 et 2 (Extracellular signal-Regulated Kinase, dénommées également 42/44) sont des enzymes phosphorylantes qui sont impliquées dans la réussite du préconditionnement. Elles sont activées par des ROS, en provenance des mitochondries, qui sortent dans le cytosol par l’ouverture du canal K+ATP. Rappelons que l’Akt est dénommée également phosphatidylinositol-3-OH kinase (PI3K) (Hausenloy et Yellon, 2004; Hausenloy et al., 2005b). Dans les cardiomyocytes, l’Akt intervient dans la protection des mitochondries en activant, par phosphorylation, l’hexokinase II mitochondriale (Miyamoto et al., 2008).
Des médicaments capables d’ouvrir le K+ATP devraient donc activer la voie du RISK. C’est le cas du Levosimendan qui était connu comme sensibilisateur au calcium et est utilisé depuis peu comme ouvreur des canaux K+ATP (du Toit et al., 2008).
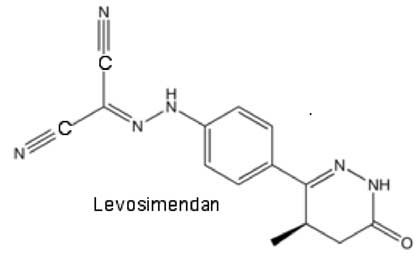
Fig. V-1 : Formule du levosimendan, ouvreur des canaux K+ATP.
B. Remise en état de l’ETC par ubiquinone exogène
L’ubiquinone Q10 possède une « mitotaxie » manifeste (Smith et al., 1999).
Molyneux et al. (2008) considèrent qu’une déficience sanguine en CoQ10 constitue un élément prédicteur de complications cardiaques prochaines.
C. Rôle de nouveaux antioxydants centrés sur les mitochondries
Malgré son tropisme mitochondrial, l’ubiquinone 10, qui fait partie de la chaîne de transport mitochondriale, a été conjuguée au triphénylphosphonium pour augmenter fortement cette « mitotaxie ». Administrée par voie buccale, elle restaure la chaîne de transport permettant la phosphorylation oxydative. Rappelons que l’usage des statines peut abaisser fortement le taux plasmatique de l’ubiquinone (Tauskela, 2007; Lowes et al., 2008).
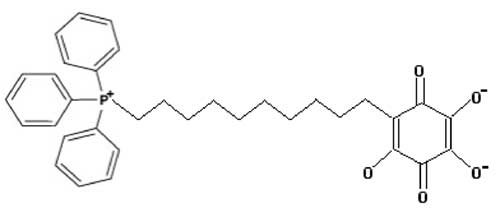
Fig. V-2 : MitoQ10 : conjugaison du CoQ10 avec le triphénylphosphonium.
D. Rôle du •NO
Kanai et al. (2001) ont indiscutablement mis en évidence la présence d’une •NO synthase dans les mitochondries cardiaques (mitNOS) ; exprimée dans le noyau, la mitNOS est adressée vers les mitochondries ; cette observation mettait fin à plusieurs années de controverses.
Le •NO généré dans les mitochondries a été impliqué dans le développement de la défaillance cardiaque (Kelm et al., 1997 ; Loke et al., 1999). Mais dès 2002, Heger et al. remettaient ces affirmations en cause en démontrant que le cœur tolère des activités élevées de la mitNOS sans conséquences fonctionnelles dangereuses. Le même groupe montra qu’une formation excessive de •NO provoquait une défaillance cardiaque chez des souris déficientes en myoglobine, tandis que la même hyperactivité, chez des souris possédant la myoglobine, était tolérée. Déjà en 2001, Flögel et al. avaient démontré le rôle protecteur de la myoglobine vis-à-vis du •NO.
Ces dernières années, le rôle des protéines antioxydantes mitochondriales (voir 1ère partie, chapitre IV et chapitre VIII) fut particulièrement étudié. Ainsi dans le cœur, le stress nitrosant conduit à la glutathionylation de protéines, ce qui amène une surproduction de peroxyrédoxines (Prxs). Cette réaction constitue un mécanisme protecteur qui s’additionne à celui de la myoglobine, mais protège mieux la mitochondrie puisque les Prxs sont aussi intramitochondriales (Reinartz et al., 2008). Les nitrites NO2- augmentent la tolérance à l’ischémie-reperfusion en amenant une véritable cytoprotection, notamment en ce qui concerne le cœur. Les nitrites diminuent l’apoptose ; cette protection s’expliquerait par la réduction des NO2- en •NO durant l’ischémie, ce qui se répercuterait sur le transport d’électrons le long de la chaîne mitochondriale, avec augmentation de la phosphorylation oxydative (Shiva et al., 2007). Cependant, en cette même année 2007, Brown et Borutaite insistaient sur l’inhibition par le •NO de la phosphorylation oxydative et de la production d’ATP, augmentant les risques d’apoptose. Ces chercheurs écrivaient en 2007 : « Dans le cœur, le •NO est produit par la •NO synthase endothéliale des cardiomyocytes… et peut-être des mitochondries… et dans des situations pathologiques, par la •NO synthase inductible dans le sarcoplasme. L’hémoglobine et la myoglobine peuvent jouer divers rôles dans la détermination des gradients d’oxygène et de •NO… Stimuler ou inhiber les •NO synthases du cœur ne produisent, in vivo, que peu de changement de la consommation d’oxygène… il reste encore peu clair si les changements observés sont dus à une inhibition directe de la respiration par le •NO, ou par une action indirecte de cet agent. »
D’autre part, Reinartz et al. (2008) affirment qu’en cas d’excès d’espèces oxygénées nitrées, il se produit une surproduction d’enzymes antioxydantes, telles la Prx VI, pour contrecarrer la nocivité de ces espèces, celles-ci réagissant par S-nitrosation de la Prx VI.
On le voit, le mode d’action protectrice des nitrites dans l’ischémie-reperfusion, qui est incontestable, est loin d’être éclairci.
Troubles de l'oxygénation et mitochondries - Sommaire |
Mitochondries et métabolisme de l'oxygène - Introduction |
Courrier |
 |
 |
 |