 Université de Liège - Centre de l'Oxygène, Recherche et Développement (CORD)
Université de Liège - Centre de l'Oxygène, Recherche et Développement (CORD) Université de Liège - Centre de l'Oxygène, Recherche et Développement (CORD)
L'Oxygène et la Vie: Tome II - L'Oxygène en Pathologie des Mammifères
Mitochondries et métabolisme de l'oxygène
Première partie : Mitochondries et oxygénation
Carol Deby
| Note: pour la facilité de la lecture, 1. chaque référence dans le texte comporte un lien vers les pages de bibliographie 2. les abréviations et formules chimiques sont reprises dans les pages du glossaire; elles sont également identifiées directement dans le texte (apparition en arrière plan lors du pointage de la souris) |
L’apoptose est considérée comme un des mécanismes biologiques les plus fondamentaux, agissant depuis le développement embryonnaire et intervenant dans les processus homéostasiques de l’adulte.
« Le rôle des mitochondries dans la régulation de la mort cellulaire est un fait qui est maintenant bien établi » (Orrenius, 2007)
« Il est devenu évident que les mitochondries participent à la mort programmée des cellules… il existe une élégante balance entre les actions opposées des Bcl-2 pro- et anti-apoptotiques. » (Chalah et Khosravi-Far, 2008)
Remarques préalables :
A. La plupart des faits rapportés ont été établis sur des modèles in vitro en l'absence de phagocytes. Mais comme l'ont souligné McHugh et Turina, en 2006, les étapes seront mieux approfondies et plus clairement distinguables en l'absence de ces cellules.
B. L'apoptose se déroule selon deux voies (Green et Evan, 2002 ; Voet et Voet, 2005) :
1. La voie intrinsèque (mort par défaut ou accidentelle) en réponse à un stress déclenché accidentellement et où les mitochondries jouent un rôle crucial.
2. La voie extrinsèque (mort sur commande ou programmée) lorsque le signal vient d'une autre cellule qui émet un peptide tel qu'une interleukine et déclenche un récepteur membranaire.
C. Les voies apoptotiques sont redondantes et, de ce fait, diffficiles à étudier (McHugh et Turina,2006).
D. L’apoptose est un moyen de défense de l’organisme, non seulement contre les proliférations cancéreuses, mais aussi contre les attaques virales. Un virus s’attaquant à une cellule pour s’y développer commence par essayer d’inhiber les voies apoptotiques de celle-ci (Best, 2008).
E. Une grande partie du programme apoptotique existe dans la cellule sous une forme latente, inactive, et ne demande qu’un stimulus apoptotique pour activer le programme et commencer l’apoptose (Krauss G, 2000).
Nous ne traiterons ici que de la voie mitochondriale ou intrinsèque, tout en rappelant brièvement et schématiquement la constitution de la voie extrinsèque.
Lectures générales conseillées : Elmore, 2007 ; Jeong et Seol, 2008.
1. Généralités sur apoptose et nécrose
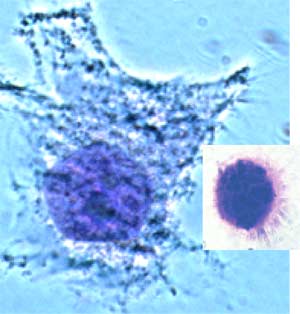
| Fig. VII-1 : Nécrose : Cellule THP-1 différenciée en macrophage; noyau normal. En médaillon, au même grossissement : THP-1 en nécrose : noyau en forme d’oursin (coloration Giemsa. Laboratoire central du CORD). |
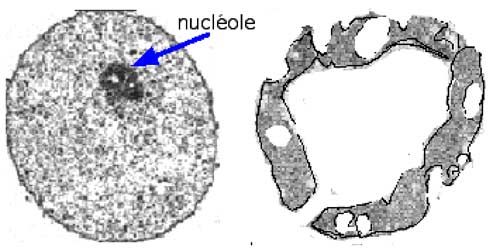
| Fig. VII-2 : Apoptose. A gauche : noyau d'hépatocyte normal ; chromatine granuleuse. A droite : noyau d'hépatocyte en cours d'apoptose. Condensation et fragmentation de la chromatine (d'après Oberhammer et al., 1993). |
La déplétion en ATP, qui est secondaire au découplage des phosphorylations oxydatives, favorise la mort de la cellule par nécrose. Durant la nécrose, le noyau se contracte et ressemble à un oursin, tandis que le cytosol se répand dans le milieu extracellulaire où il sera phagocyté, tout comme le noyau.
L'apoptose se déroule en plusieurs étapes comprenant des cascades d'activation d'enzymes aboutissant à l’activation d’endonucléases qui scindent le DNA en plusieurs fragments. Morphologiquement, la première caractéristique de la cellule apoptotique est la fragmentation irrégulière de la masse nucléaire qui a fait croire aux hématologues du début du XXième siècle à l'existence de plusieurs noyaux dans certains globules blancs (les "polynucléaires").
Biochimiquement, la différence majeure entre nécrose et apoptose est que l'apoptose nécessite de l'énergie sous forme d'ATP et non la nécrose (Halestrap, 2006).
2. Les 2 types d'apoptose
voir Chalah A, Khosravi-Far R. The mitochondrial death pathway. Adv Exp Med Biol. 2008; 615: 25-45.
A. Généralités
Presque tous les processus apoptotiques sont réalisés par une famille de protéases intracellulaires : les caspases, protéines à cystéine caratérisées par des aspartates dont le rôle est catalytique (Cystéine-ASPartate). Les caspases sont synthétisées en tant que zymogènes inactifs, les procaspases inactives. Un activateur vient cliver une première procaspase au niveau d’un aspartate. La caspase active se sépare ainsi du résidu cystéinyl qui l’inactivait et va, à son tour, libérer une deuxième caspase qui agira de même sur une troisième. Une cascade de protéases agissent ainsi jusqu’à activer des DNAases nucléaires qui mettront en pièces le DNA nucléaire, produisant les fragments nucléosomiques, tandis que la membrane cellulaire se réduit en une structure bulleuse et que la cellule apoptotique se résout en petites vésicules : les apoptosomes (Alnemri et al.,1996 ; Wyllie et al., 1984 ; Cain, 2003 ; Yang et al.,1998 ; Salvesen et Dixit, 1997).
Les caspases des mammifères sont classées en :
• initiatrices : Casp-9, Casp-2, Casp-8, et Casp-10
• effectrices : Casp-3, Casp-7, Casp-6 (Kumar et Dorstyn, 2009)
B. L'apoptose physiologique ou extrinsèque
L'apoptose physiologique est la mort d’une cellule programmée physiologiquement par le plan de développement embryologique ou exigée pour le maintien de la forme. Ce processus, sous contrôle génétique, s'est conservé au cours de l'évolution et est indispensable pour le développement de l'embryon, puis pour la conservation morphologique de l'adulte et la lutte contre une multiplication anarchique des cellules des couches génératrices.
C. L'apoptose accidentelle ou intrinsèque
Elle est pathologique et vient de la mitochondrie après une lésion de la cellule ou une situation pathologique, telle qu’une anoxie.
3. L'apoptose extrinsèque
Dans cette forme physiologique, le processus apoptotique ne met pas (ou très peu) en cause les mitochondries. Nous nous bornerons à rappeler brièvement le schéma de la cascade apoptotique physiologique.
Le signal mortifère vient de l'extérieur. Décrivons un mécanisme devenu classique : les macrophages sont producteurs de FAS ligand (FAS L), protéine à laquelle correspond, sur des cellules cibles visées par ces globules blancs, une protéine membranaire fixatrice : le récepteur de mort (death receptor) ou FAS (Figure VII-3 A-C). Le Fas-ligand le plus célèbre est certainement le TNF-α. Le complexe FAS/Fas ligand recrute dans le cytosol de la cellule condamnée des protéines d'adaptation (adaptor ou FAS-associated-death-domain : FADD) fixant la pro-caspase-8 et, en la clivant, lui fait perdre le résidu cystéinyl : elle devient ainsi la caspase-8 active.
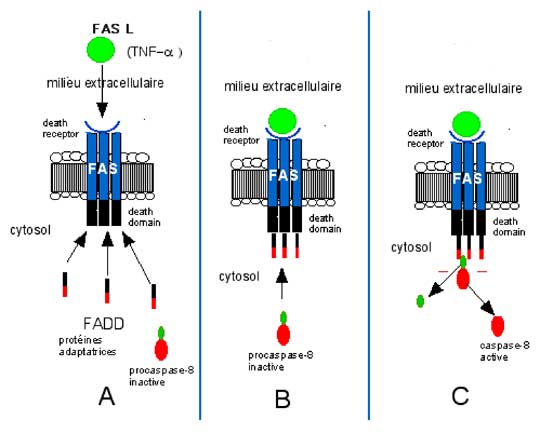
| Fig. VII-3 A-C : Cascade d’activation de l’apoptose extrinsèque. NB : Avant que le FAS L ne se lie au FAS, les 3 éléments identiques de celui-ci sont relativement épars dans la membrane ; c’est lors de la formation du complexe FAS/FASL (B) que la trimérisation s’effectue, suivie d’un appel de protéines adaptatrices pour former le FADD ; ce dernier attire une procaspase-8 qui est activée et libérée dans le cytosol (C). |
La caspase-8 s’attaque à la procaspase-3 qui à son tour devient active (Figure VII-3 D). Une avalanche de phénomènes protéasiques se termine dans le noyau avec l’activation d'une DNAase qui attaque le DNA nucléaire qui se rompt en gros morceaux.
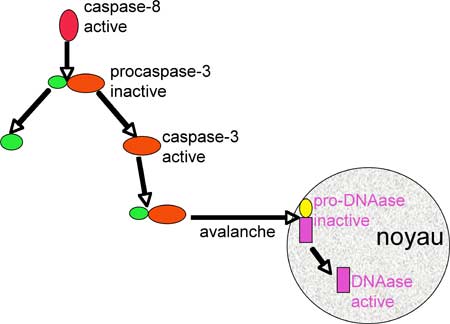
| Fig. VII-3 D : La caspase-8 (initiatrice) libère la caspase-3 (effectrice) qui provoque une avalanche d’activations aboutissant à celles de DNAases nucléaires. |
• Note sur les récepteurs Fas
Ce sont des protéines transmembranaires de 335 acides aminés, pesant 50 kDa, qui, après fixation de leurs ligands par leur extrémité N-terminale, sont activées par rassemblement sous forme de trimères. L'extrémité intracellulaire C-terminale comporte une séquence de 80 acides aminés formant un "domaine de mort" (Death Domain ou DD). Dans le cas du Fas, les trois DD associés du trimère Fas activé vont recruter une protéine cytoplasmique FADD (Fas Associating protein with Death Domain) comportant deux domaines: un domaine DD qui forme un dimère avec le domaine DD de Fas et un domaine DED (Death Effector Domain) qui fixe et active la procaspase 8 (forme zymogène inactive de la caspase-8). En activant la procaspase 8, le Fas induit la cascade d'activations protéiques aboutissant à la mort cellulaire par apoptose. Le Fas constitue la voie extrinsèque de déclenchement de l'apoptose.
• Fas ligand (FasL)
Il provient d’autres cellules, surtout des macrophages. Il va induire l’apoptose dans des cellules condamnées.
Un exemple fameux est le TNF (tumor necrotizing factor), une cytokine qui est secrétée par les macrophages et certaines cellules immunologiquement compétentes et se lie au récepteur du Fas pour former le complexe mortifère Fas/FasL.
4. L'apoptose intrinsèque
Elle dépend étroitement de la mitochondrie
Dans les situations pathologiques telles que l'ischémie-reperfusion et le stress oxydant par exemple, la mitochondrie est considérée comme l'organelle provoquant la mort cellulaire (Orrenius et al, 2007).
Le phénomène de départ est l'accroissement de la MOMP (Mitochondrial Outer Membrane Permeability) ou mPT pouvant aboutir à la rupture de cette membrane externe (Green et Kroemer, 1998). Certaines protéines situées dans les espaces intermembranaires se répandent alors dans le cytosol (Green et Kroemer, 2004 ; Brookes et al., 2004).
A. Mécanisme général : la mPT (mitochondrial Permeability Transition)
Une bonne introduction historique est fournie par la revue de Bernardi et al., 1999.
1. Historique du concept
En 1979, Haworth et Hunter décrivirent l'existence, in vitro, d'augmentations abruptes de la perméabilité de la membrane mitochondriale interne en réponse au Ca2+, aux stress oxydants et à la déplétion en ATP (Haworth et Hunter, 1979). La perméabilité transitoire de la mitochondrie (mPT) est un accroissement soudain de la perméabilité de la membrane mitochondriale interne pour l'eau et des molécules de taille inférieure à 1,5 kDa. Des recherches intensives menèrent à la conception d'une vanne ou pore large (mégapore), de nature protéique, non sélectif, inhibé par la cyclosporine A et excité par le Ca2+ (Crompton et al, 1988; Crompton et Costi 1988; Crompton, 1999). Ce mégapore est désigné sous le sigle mPTP (mitochondrial Permeability Transition Pore) (Weiss et al., 2003). L’inhibition du mPTP par la cyclosporine est étroitement dépendante de la présence de phosphate (Basso et al., 2008). D’autres régulateurs du mPTP sont constitués par des analogues de l’ubiquinone, certains étant inhibiteurs, d’autres activateurs (Walter et al., 2000).
La réduction des disulfures au niveau des zones mPTP peut protéger celles-ci contre l’ouverture du pore (Wudarczyk et al., 1996).
Ce soudain accroissement de la perméabilité de la membrane interne aux ions et aux substances dissoutes produit une dissipation du gradient électrochimique des protons ΔΨm (Bernardi, 1992), une perte de l'homéostasie ionique (Basso et al., 2005) et un déséquilibre de la balance redox en faveur de l’oxydation (Bindoli et al., 1997; Jones, 2006).
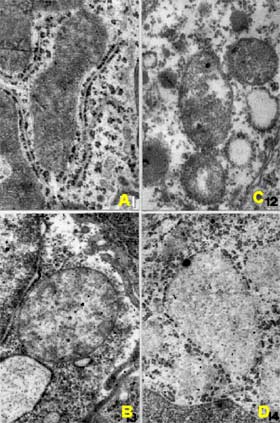
| Fig. VII-4 : Tissus de la sous-muqueuse intestinale de souris irradiées par radiations X de 250 kV à dose létale (750 rad). A : mitochondrie de souris témoins. B, C, D : tissus prélevés 2 heures, 8 heures et 12 heures après irradiation. Un "swelling" progressif arrondit l'organelle dont les crêtes disparaissent progressivement. Après 12 heures, les structures mitochondriales ont disparu (Carol DEBY et Kanaya MALKANI, 1969, Laboratoire de Physique de l’Université d'Alger et Institut Pierre et Marie Curie, Alger). |
2. Causes de la mPT
Ce phénomène est produit par l'ouverture du pore non spécifique mPTP au cours d'une accumulation d'ions calciques (Halestrap et al., 2002).
- accumulation d'ions calciques, due à des phénomènes toxiques comme le peroxynitrite (Borutaite et al., 1999) ; les mitochondries peuvent accumuler le Ca2+ jusqu’à un certain point de saturation qui est abaissé lors de phénomènes oxydants (Orrenius et al.,. 2007). Pour le rôle du mitK+Ca (O'Rourke et al, 2005), voir chapitre V.
- stress oxydants (particulièrement durant l'anoxie-réoxygénation)
- irradiations ionisantes (rayons X, gammas et UV, générateurs de ROS).
3. Conséquences de la mPT : hauts risques d’apoptose
Diverses substances apoptogènes sont libérées dans le cytosol, dont principalement :
- le cytochrome c, faisant normalement partie de la chaîne respiratoire,
- l'AIF (Apoptosis-inducing factor).
4. Effets physicochimiques de la mPT (détails dans Quinlan et al., 2008)
Lorsque la durée de la transition de perméabilité est trop longue, des troubles graves se produisent :
1) Découplement de la phosphorylation oxydative avec chute de la synthèse d'ATP.
2) Dissipation du gradient électrochimique des protons ΔΨm (Bernardi, 1992) avec une perte de l'homéostasie ionique (Basso et al, 2005).
3) Hydrolyse de l'ATP et aggravation de la carence en ATP.
4) Altération de la membrane externe et libération de cytochrome c dans le cytoplasme et constitution d’apoptosomes (Bernardi, 1999).
5) Mort par nécrose de la mitochondrie (voir les parties C et D de la figure VII-4 ci-dessus).
6) Induction de l'apoptose cellulaire.
B. Explications de la mPT
De nombreuses recherches furent entreprises dès la date de la découverte de la mPT. Trois alternatives s’offraient :
1. La mPT se produit à l’occasion d’une déchirure de membranes mitochondriales.
2. De véritables canaux existent, ne s’ouvrant que lors de situations d’urgence. Plaide en faveur de leur existence le fait que l’hyperperméabilité peut être transitoire et ne mène pas nécessairement à la mort cellulaire.
3. La coexistence des deux processus, déchirure et structure protéique dormante, est possible
1. Déchirure de la membrane mitochondriale externe
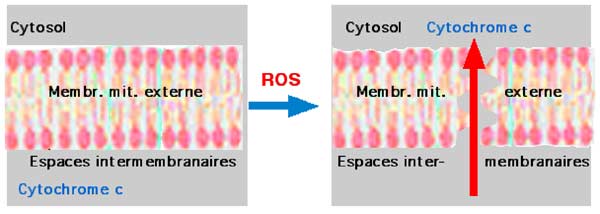
Fig. VII-5 : Hypothèse de la déchirure
2. Ouverture d'un pore ou d'un canal virtuel dans la membrane externe
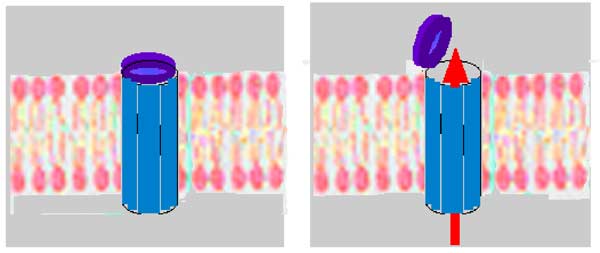
| Fig. VII-6 : Hypothèse du pore géant normalement fermé. à gauche : situation normale, le pore est clos ; à droite : le pore est ouvert et permet la sortie de molécules de taille inférieure à 1,5 kDa, par action de Ca2+ ou de ROS ouvrant le pore. |
3. Coexistence des deux processus
Les deux phénomènes peuvent se produire : dans le cas du pore, la situation peut être réversible ; c’est sur cette notion que les cliniciens pratiquent maintenant le préconditionnement. La déchirure membranaire, elle, mène à l’apoptose.
C. Développement de la théorie du pore mPTP (mégapore)(mitochondrial Permeability Transition Pore).
Voir l’excellent article de revue de Bernardi et al., 2006.
1. Caractéristiques
Il s’agit d’un pore géant (mégapore) non spécifique, laissant passer des molécules de taille inférieure à 1,5 kDa (Halestrap et al., 2002). Ce pore est associé au phénomène de transition de perméabilité mitochondriale, la mPT. En s'ouvrant, il permet des échanges non spécifiques entre la matrice et l'espace intermembranaire. Normalement, ses ouvertures ne peuvent être que très transitoires ; ce pore semble jouer un rôle de soupape de sécurité contre des élévations de très courte durée de la concentration de divers ions, surtout Ca2+ (Crompton, 1999).
Le mPTP est un acteur central dans la mort cellulaire dépendant de la mitochondrie, en libérant dans le cytosol des substances proapoptotiques :
- le cytochrome c
- l'AIF.
In vitro, l'ouverture de ce pore diminue la force protonomotrice et provoque divers troubles :
- effondrement du ΔΨm,
- arrêt de la synthèse d'ATP d'où décroissance de l'ATP intracellulaire (Imberti et al., 1993; Ankarcrona et al., 1995),
- accroissement du taux cytosolique de Ca2+ (Imberti et al., 1993; Pastorino et al., 1995),
- gonflement mitochondrial (swelling) (Figure VII-4),
- chute de la force libérée par la pompe à protons (Fontaine et al.,1998; Dumas et al., 2009).
2. Structure
Le mégapore est formé d'au moins 3 éléments (Crompton, 1999 ; Weiss et al., 2003; Basso et al., 2005; Halestrap et al., 2004) :
1. Le canal anionique dépendant du voltage (Voltage-Dependent Anion Channel : VDAC).
2. La translocase des nucléotides adényliques (Adenine Nucleotide Translocase : ANT), traversant la membrane interne.
3. La cyclophiline-D (CyP-D) protéine matricielle, inhibitrice de l’ouverture du pore (Schinzel et al., 2005).
4. L’hexokinase et la créatine kinase sont souvent citées, mais pas unanimement (Beutner et al., 1998).
5. Il est certain que les protéines pro- et anti-apoptotiques dont Bcl-2 et Bax jouent un rôle dans le fonctionnement du pore (Brenner et al., 2000).
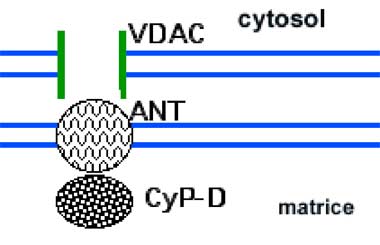
Fig. VII-7 : Les 3 éléments classiques du mPTP.
3. Déclenchement de l'ouverture du mPTP
Comme l'avaient bien observé ses découvreurs, ce pore s'ouvre sous l'effet de haute concentrations de Ca2+ dans la mitochondrie, mais aussi par stimulation par les ROS. La déplétion en nucléotides d'adénine agit de même (Halestrap et Brenner, 2003).
4. Modalités et facteurs de l'ouverture du mégapore
On a pensé longtemps que cette ouverture était irréversible et donc que la mort était imparable.
Mais le préconditionnement (que nous verrons plus loin) a montré que ce phénomène pouvait être interrompu. Cette réversibilité, déjà supposée par les pionniers des recherches sur le mPTP, Haworth et Hunter, en 1979, est chose admise en 2009 (Dumas et al., 2009).
L'ouverture du mégapore est soumise à divers facteurs (Bernardi et al., 1994; Gunter et al., 1994; Zoratti et Szabò, 1995; Halestrap et al., 2004);
a. La différence de potentiel transmembranaire ΔΨm : la décroissance de ΔΨm favorise l’ouverture du pore;
b. le pH matriciel, lorsqu’il s’abaisse en dessous de 7,0;
c. le potentiel redox;
d. l'abaissement de concentration des nucléotides adényliques, tels que NADH;
e. la concentration en cations bivalents tels que Ca2+;
f. les ROS (voir plus bas);
g. divers agents pharmacologiques (voir ci-dessous, point 6) ; rappelons que le plus ancien de ceux-ci est la cyclosporine A, inhibitrice de l'ouverture du mégapore (Broekemeier et al., 1995; Woodfield et al., 1998 ; Hansson et al., 2004).
L'ouverture du mPTP peut être réversible (voir paragraphe suivant), mais toutes ses conséquences ne le sont pas. La fermeture restaure la force de la pompe à proton et l'homéostasie des ions se rétablit. Mais les molécules qui ont anormalement traversé la membrane (par exemple le cytochrome c) resteront prisonnières à leur place anormale, à l'intérieur ou à l'extérieur de la mitochondrie.
5. Les deux états du mégapore in vitro
Dans les mitochondries isolées, deux états sont observables au niveau du mégapore (Ichas et Mazat, 1998):
a. Un état de faible conductivité réversible (ou ouverture transitoire) conduisant à la diffusion d'ions de petite taille, tels Ca2+, dépendant du pH et déclenchant la fermeture du pore. Cette situation protègerait les cellules d'une lésion irréversible en évitant le « swelling » et la libération dans le cytosol de facteurs pro-apoptotiques tels que le cytochrome c (Huser et Blatter, 1999 ; Ichas et al., 1997 ; Ichas et Mazat, 1998 ; Hausenloy et al., 2004). Un tel mode transitoire et l’ouverture suivie de fermeture peuvent être répétés, par exemple, avec de très faibles concentrations de peroxyde d'hydrogène (± 2 μM) sur coeur isolé et perfusé par une solution physiologique dans laquelle on réalise pendant un temps court une concentration déterminée d’H2O2 (Saotome et al., 2009). Durant cette ouverture de faible conductance, certains ions en excès, tels le Ca2+, sortent de la mitochondrie et peuvent inhiber l'installation d'une phase de haute conductivité (Huser et Blatter, 1999; Ichas et al., 1997; Ichas et Mazat, 1998 ; Hausenloy et al., 2004).
b. Un état de haute conductivité irréversible (ouverture permanente), aboutissant à la diffusion non sélective de molécules plus grosses (>1,5kDa) telles que le cytochrome c; cette situation ouverte peut se stabiliser, amenant l’effondrement définitif de ΔΨm, suivi de la destruction des structures mitochondriales, libérant des facteurs provoquant l'apoptose cellulaire (Halestrap et al., 2004 ; Ichas et Mazat, 1998 ; Zorov et al., 2009).
6. Inhibiteurs du mPTP
l’atractyloside (Moret et al., 1966)
l’acide bongkrekique (Henderson et Lardy, 1970)
7. Séquence des événements après ouverture du mégapore en état de haute conductivité.
a) Gonflement caractéristique de la matrice (« swelling ») (Petit et al., 1998) (figure VII-4)
b) Effondrement du ΔΨm (Ly et al., 2003)
c) Arrêt de la synthèse d'ATP.
d) Hydrolyse de l'ATP.
e) Décroissance du taux intracellulaire d'ATP (Imberti et al., 1993; Ankarcrona et al., 1995).
f) Fuite dans le cytoplasme de petites molécules dont le cytochrome c (Liu X et al., 1996; Green, 2000).
g) Accroissement du taux cytosolique de Ca2+ (Imberti et al., 1993; Pastorino et al., 1995).
h) Sortie dans le cytosol d'une caspase mitochondriale pré-apoptotique.
i) Chute de la force libérée par la pompe à protons (Fontaine et al., 1998; Dumas et al., 2009).
En 2009, elle fait toujours l'objet de débats (Dumas et al., 2009)
1. Conception multiprotéique
Le mPTP serait formé d'au moins 3 éléments, comme nous l’avons vu ci-dessus (Crompton, 1999; Weiss et al., 2003; Basso et al., 2005; Halestrap et al., 2004).
Une conception plus compliquée a été proposée par Barrett et al. (2006), comprenant, outre les 3 protéines énumérées sur la figure VII-7, une hexokinase et une créatine kinase.
D'autres protéines constitutives auraient été isolées, dont une créatine kinase (CK).
2. Rôle régulateur des protéines de la famille protéique cytosolique des Bcl-2
a) Bcl-2 anti-apoptotiques : Bcl-2 et Bcl-XL ralentissent le mécanisme de déclenchement de l’apoptose ou l’annihilent, permettant la survie de la cellule.
b) Bcl-2 pro-apoptotiques : Bax, Bad et Bak déclenchent la perméabilité cellulaire transitoire, mais nécessitent la présence du canal anionique VDAC, ce qui démontre la nécessité de cette protéine dans la structure du mPTP (Yamagata et al., 2009). Le VDAC peut interagir avec l’hexokinase pour empêcher son activité anti-apoptotique.
3. Controverses en 2009
A ce jour, on n'en connaît pas plus sur la composition exacte du mPTP (Zorov et al., 2009). Il avait même semblé que l'ANT et la cyclophiline n'entraient pas dans la structure du mégapore, mais seraient uniquement des agents régulateurs. Toutefois, des travaux récents ont montré que, dans le pré- et le post-conditionnement, l'absence de Cyp-D rendait les traitements inopérants (Lim, 2007). Déjà en 2005, Baines et al. avaient démontré que la perte de cyclophiline D avait mis en évidence son rôle dans la mPT. La même équipe avait montré, en 2007, le rôle pré-éminent du VDAC dans l’apoptose cellulaire (Baines et al., 2007).
E. Rôle des facteurs mitochondriaux pro-apoptotiques
Nous avons vu ci-dessus que l’ouverture du mégapore permettait la sortie dans le cytosol de deux facteurs proapoptotiques, le cytochrome c et l’AIF. Deux voies principales mènent à l’apoptose :
• la voie des caspases,
• la sortie de l‘AIF des mitochondries.
1. Voie des caspases (Green, 2000; Orrenius et al., 2007; Hand et Menze, 2008)
Le cytochrome c, qui fait partie de l’ETC, est libéré du lien le rattachant à une cardiolipine de l’espace intermembranaire, par la peroxydation de cette dernière (Tuominen et al., 2002 ; Fariss et al., 2005). Le mécanisme de cette peroxydation est étudié plus bas (paragraphe F). L’hème subit alors une translocation qui le fait sortir par le mégapore dans le cytosol où il rencontre une grosse protéine cytosolique (130 kDa), l’Apaf-1 (Apoptotic Protease Activating Factor 1) (Liu X et al.,1996 ; Zou et al., 1997).
• L’Apaf-1 consiste en trois domaines fonctionnels (figure VII-8) :
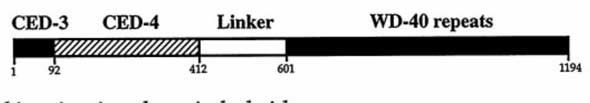
Fig. VII-8 : Schéma de la constitution d’une molécule d’Apaf-1

Fig. VII-9 : Partie répétitive WD-40, formée de 40 aminoacides, terminée souvent par un dipeptide Trp-Asp (Adrain et al., 1999).
En se liant à l’Apaf-1, le cytochrome c provoque une série de transformations nécessitant la présence de désoxy-ATP (d-ATP) : figure VII-10 (Liu X et al., 1999; Zou et al., 1999).
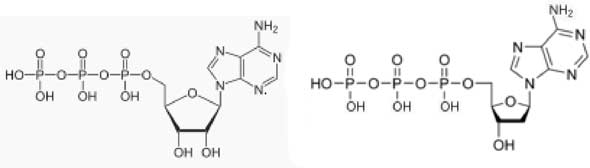
Fig. VII-10 : à gauche : l'ATP ; à droite, le désoxyATP (dATP).
Le dATP joue donc le rôle de cofacteur.
L’activation continue par l’oligomérisation de 7 complexes Apaf-1-cytochrome c qui s’unissent de manière radiaire (Figure VII-11) pour former l’apoptosome, grand complexe d'environ 1400 kDa (Srinivasula et al., 1998; Cain, 2003; Kim et al., 2005). Cette structure s’organise autour des sept régions répétitives WD40 (Adrain et al.,1999; Acehan et al., 2002).
La liaison du cytochrome c à l’Apaf-1 provoque l’hydrolyse du dATP en dADP qui est par après remplacé par 1 dATP exogène. L’hydrolyse du dATP et l’échange, sur l’Apaf-1, sont deux étapes indispensables pour la formation de l’apoptosome (Kim et al., 2005).
• L’apoptosome : structure de départ pour l’activation de la caspase 9
Pour Kumar et Dorstyn (2009), l’apoptosome jouerait un rôle équivalent au complexe protéique d’adaptation qui active la caspase-8 dans la voie extrinsèque (Bao et Shi, 2006).
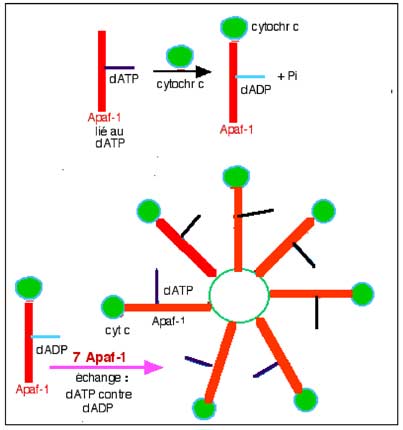
| Fig. VII-11 : La protéine Apaf-1 se lie d'abord à une molécule de déoxyadénosine triphosphate, dATP, pour réagir avec le cytochrome c pour former un complexe Apaf-1/cytochrome c/dADP, avec libération d'une molécule de phosphate Pi (partie supérieure de la figure). En présence de dATP, qui s'échange avec le dADP, 7 complexes s'assemblent pour former une structure rotacée active : c'est l'apoptosome actif. Sans le dATP, des assemblages de 7 complexes de structure non rotacée se forment : apoptosomes inactifs. Le cercle vert délimite les zones d’accrochage des 7 terminaisons répétitives WD-40 (Li et al., 1997; Kim et al., 2005). |
Cet apoptosome, complexe protéique activant les procaspases-9, capte une molécule de celles ci pour former un complexe enzymatique actif. Le complexe activera ensuite une procaspase-3 ; la caspase-3 clivera la procaspase-7, initiateur de cascade de caspases et d’autres agents protéasiques, aboutissant à l’activation de DNAases nucléaires (Yang et al., 1998).
2. Sortie de l'AIF (Apoptosis-Inducing Factor)
L'Apoptosis-Inducing Factor (AIF) est un facteur induisant l'apoptose mitochondriale; il dépend de l'état rédox (Churbanova et Sevrioukova, 2008). A la fin de la décennie 1990, l’AIF fut purifié et identifié comme élément clé dans la mort cellulaire indépendante des caspases : les inhibiteurs des caspases n'inhibent pas l'apoptose induite par l'AIF (Susin et al., 1999). La protéine pro-apoptotique Bax n’affecte pas la sortie du cytochrome c, mais inhibe celle de l’AIF, sur mitochondries isolées (Arnoult et al., 2002).
Dans les conditions physiologiques, l'AIF est situé dans la mitochondrie où, par ses propriétés d'oxydoréductase (Miramar et al., 2001), il intervient dans le transport d'électrons et par là, dans la phosphorylation oxydative (Vahsen et al., 2004).
C'est une flavoprotéine qui provoque l'apparition de certaines caractéristiques de l'apoptose dans les cellules vivantes lorsque sa forme recombinante est injectée en quantités physiologiques : la chromatine se condense dans les noyaux, le DNA est dégradé en morceau de 50 kilobases (Ye et al., 2002).
Après une agression, sous l'effet d'un stimulus apoptogène, l'AIF est clivé par une calpaïne ou par une cathepsine dans sa localisation intramitochondriale (Polster et al., 2005; Yuste et al., 2005).
Après l'ouverture du mégapore, l'AIF pénètre dans le cytosol et est transporté dans le noyau après un clivage par protéase (Polster et al., 2005) où il attaque le DNA qui se fragmente en grands morceaux, sans intervention de caspase, tandis que le potentiel transmembranaire des mitochondries disparaît (Qiu et al., 2008). L’AIF est la première protéine identifiée dans la voie apoptotique indépendante des caspases (Susin et al., 1999; Daugas et al., 2000).
L'AIF est synthétisé et libéré dans le cytosol sous forme d’un précurseur de 67 kDa, importé et localisé dans la mitochondrie où le prodomaine est enlevé laissant la forme mature de 57 kDA ; l’AIF est confiné dans l'espace intermembranaire (Daugas et al., 2000).
L’AIF comprend trois domaines :
• la région fixant le FAD
• la région fixant le NADH
• le domaine contant la partie porteuse du COOH terminal
Les deux premiers domaines constituent la partie assimilable à une oxydo-réductase (Lorenzo et Susin, 2004), car elle peut transférer un électron et joue un rôle dans la phosphorylation oxydative.
Cette flavoprotéine exerce donc une double fonction :
a) dans la mitochondrie normale, elle intervient dans la phosphorylation oxydative (Miramar et al., 2001; Vahsen et al., 2004) ainsi que dans la régulation redox (Daugas et al., 2000; Qiu et al., 2008).
b) elle joue un rôle capital dans de nombreux processus apoptotiques, sans faire intervenir la voie du cytochrome c (Arnoult et al., 2002). Mais elle pourrait jouer un rôle anti-apoptotique via ses propriétés de peroxydase. 100 µM de peroxyde d'hydrogène provoquent la surexpression de l'AIF in vitro (Qiu et al., 2008).
En résumé, c'est une protéine bifonctionnelle, ayant d'une part un rôle essentiel en bioénergétique (dans la mitochondrie), et d'autre part une fonction vitale en intervenant dans l'apoptose (lorsque l'AIF pénètre dans le noyau). Les deux activités (oxydo-réductase et pro-apoptotique) sont dissociables. Les deux activités, celle d'oxydo-réductase et celle de DNAase, peuvent être séparées (Miramar et al., 2001; Delettre et al., 2006).
F. Rôle du complexe cytochrome c/cardiolipine
Une série de découvertes, durant le début de ce siècle, ont mis en évidence le rôle capital joué par l’association cytochrome c/cardiolipine (cyt c/CL). En 2008, Houtkooper et Vaz n’ont pas hésité à titrer un article : Cardiolipin, the heart of mitochondrial metabolism. Nous avons déjà donné sa formule dans le chapitre II. La cardiolipine est indispensable au maintien de la structure quaternaire et des fonctions du complexe III (Gomez et Robinson, 1999). Cinq CL sont liées au cyt c (Kagan et al., 2009b) ; elle est aussi indispensable au fonctionnement du complexe IV auquel sont associées 2 molécules de CL (Robinson et al., 1990). Une molécule de complexe V est associée à 4 molécules de CL (Eble et al., 1990). Mais le fait le plus marquant est que le complexe cyt c/CL peut acquérir, par un changement de conformation du cyt c, une activité peroxydasique spécifique (Kagan et al., 2005), d’autant plus facilement que les acides gras de la CL sont insaturés. Cette peroxydase affecte lentement la destruction d’H2O2, mais l’hème passe par les stades classiques d’oxydation, notamment celui de l’ion ferryl, créateur de radicaux libres, pouvant arracher des atomes H• sur les chaînes insaturées de la cardiolipine du complexe (voir Initiation au métabolisme de l’oxygène, chapitre VIII) : un cycle d’autoxydation est initié et le cytochrome c est libéré, pouvant être expulsé dans le cytosol. Un fait remarquable est que l’activité peroxydasique est liée au complexe cyt c/CL (Kagan et al., 2005).
5. Intervention des ROS dans l'apoptose (Wang, 2001; Orrenius, 2007)
Si les ROS (voir chapitre VI) jouent un rôle prééminent dans diverses pathologies ; ils interviennent dans des mécanismes physiologiques, notamment dans la transmission des signaux où ils peuvent agir comme seconds messagers (Nakamura et al., 1997).
La majorité des ROS sont produites dans les mitochondries (Orrenius, 2007).
Un des effets délétères potentiels des ROS est la facilitation d’apparition de la perméabilité transitoire mitochondriale (mPT) dépendante du Ca2+.
Dumont et al. (1999) ont démontré qu'au-dessus d'une certaine concentration d'H2O2 dans le milieu extracellulaire (>10-5M), on pouvait indiscutablement mettre en évidence dans les cellules des signes d'apoptose intrinsèque comme le clivage de la caspase-3, due à l'ouverture de mégapores, qui est confirmée par les signes classiques : libération de cytochrome c dans le cytosol et abaissement du potentiel transmembranaire ΔΨm. La contre-épreuve, consistant à inhiber l'ouverture du mégapore par emploi de l'acide bongkrekique, inhibe l'apoptose provoquée par H2O2.
La concentration en H2O2 réalisée intracellulairement par une concentration extracellulaire de 10-4M est dix fois moindre (Song et al., 2007). Ce sont donc des concentrations de 10-5M qui provoquent l'apoptose. De 10-9M à 7.10-7M, le peroxyde d'hydrogène se comporte comme un second messager non létal (Song et al., 2007).
◊ Rôle des lipoperoxydes : les lipoperoxydes altèrent les fonctions mitochondriales vitales, telles que l’ETC, la phosphorylation oxydative et le maintien du potentiel de membrane de la mitochondrie.
◊ ROS et libération du cytochrome c : la cardiolipine n’est présente que dans les mitochondries dont elle assure la fluidité des membranes. Des interactions moléculaires se produisent par insertion d’une chaîne grasse de la CL dans un canal hydrophobe sis dans le cytochrome c (Tuominen et al., 2002). Des modifications oxydatives modifient la solidité de cette liaison (Ott et al., 2002; Paradies et al., 2000).
Kagan et al. (2005) ont montré qu’une peroxydation sélective de la CL précède la libération du cytochrome c durant l’apoptose. L’oxydation est catalysée par une activité peroxydasique spécifique de la cardiolipine liée au cytochrome c (Kagan et al., 2005; Bayir et al., 2006; Kagan et al., 2004; Jiang et al., 2003; Kagan et al., 2006; Belikova et al., 2006).
A. Famille des Bcl-2 : pro et anti-apoptotique : voir D.2.a
B. Effets de la décroissance de l'ATP
La diminution de la phosphorylation oxydative s'accompagne d'une hausse de la génération de ROS avec les effets cyotoxiques que nous décrivons ci-dessus (Orrenius, 2007).
C. Enzymes antioxydantes
L’apoptose due à un excès de ROS est régulée par les enzymes antioxydantes (voir chapitre VIII).
Mitochondrie et oxygénation - Chapitre VII:Protection anti-ROS mitochondriale |
Troubles de l'oxygénation et mitochondries - Sommaire |
Mitochondries et métabolisme de l'oxygène - Introduction |
 |
 |
 |