Université de Liège - Centre de l'Oxygène, Recherche et Développement (CORD)
Université de Liège - Centre de l'Oxygène, Recherche et Développement (CORD) Université de Liège - Centre de l'Oxygène, Recherche et Développement (CORD)
L'Oxygène et la Vie: Tome II - L'Oxygène en Pathologie des Mammifères
Mitochondries et métabolisme de l'oxygène
Seconde partie : Troubles de l'oxygénation et mitochondries
Carol Deby
| Note: pour la facilité de la lecture, 1. chaque référence dans le texte comporte un lien vers les pages de bibliographie 2. les abréviations et les formules chimiques sont reprises dans les pages du glossaire ; elles sont également identifiées directement dans le texte (apparition en arrière plan lors du pointage de la souris) |
Les observations réalisées sur cultures cellulaires ont montré des différences nettes entre la phase anoxique et celle de réoxygénation, aussi bien morphologiques que biochimiques.
"Mitochondria are increasingly recognized as lynchpins in the evolution of cardiac injury during ischemia and reperfusion" (Chen Q et al., 2007)
C’est la situation que subiront des tissus irrigués par une artère terminale où la circulation est interrompue soit par manœuvre expérimentale (clampage de l’artère), soit par un mécanisme pathologique (spasme de l’artère ou obstruction par un caillot) (voir introduction, figure 1).
Les travaux dans ce domaine ont été centrés principalement sur le myocarde. Au niveau de cet organe, une ischémie expérimentale de moins de 20 minutes est suivie d’une reperfusion sans dommages, tandis que plus de 45 minutes d’anoxie entraînent inévitablement les lésions dites d’ischémie reperfusion (Park et Lucchesi, 1999).
On a admis difficilement que c’est durant la reperfusion, phase évidemment incontournable, que se produisent les lésions graves (Kloner, 1993). A la fin du siècle dernier, le doute planait toujours (Ferrari et Hearse, 1997). Pourtant les recherches ultérieures ont clairement établi que les lésions des myocytes n’apparaissent que lorsque la réoxygénation est rétablie (Park et Lucchesi, 1999).
Une définition rigoureuse du phénomène a été fournie par Rosenkranz et Buckberg (1983) :
« une conséquence métabolique, fonctionnelle et structurelle du rétablissement de la circulation coronaire qui peut être contrecarrée par des modifications des conditions de reperfusion ».
Cette définition ouvrait un champ d’espoir qui se matérialiserait 3 ans plus tard (voir chapitre IV).
Tous les chercheurs reconnurent le rôle essentiel exercé par les mitochondries, une atteinte de celles-ci amenant la perte de viabilité de la cellule (Di Lisa et Bernardi, 2006). De nombreux travaux vinrent confirmer l’importance des mitochondries dans le mécanisme des troubles de la reperfusion.
Des doutes subsistaient quant au déroulement des opérations pathologiques, mais un consensus s’établit autour du rôle crucial de l’ouverture du mPTP, un peu négligée par Petrosillo et al. (2004).
La dysfonction mitochondriale commença à être décrite comme suit :
On constate d’abord une altération du fonctionnement du complexe I (CoI) dans les 10 à 20 premières minutes de l’ischémie (Flameng et al., 1991; Rouslin, 1983) avec atteinte des cardiolipines (Paradies et al., 2004) ; puis les autres éléments de l’ETC sont atteints et la production d’ATP s’effondre (Lesnefsky et al., 2004a et b). Le dysfonctionnement de CoI entraîne, par diminution du nombre d’électrons transportés, une hausse temporaire des ROS formés (Turrens et Boveris, 1980). Le CoIII est lésé par atteinte des protéines à Fe-S ; dans les mitochondries de cœur de rat, l’ischémie-reperfusion provoque une nette diminution de l’activité du CoIII, tandis qu’une chute en cardiolipine est constatée, provoquée par lipoperoxydation (Petrosillo et al., 2003a). Le CoIV des mitochondries du myocarde est modifié par le 4-hydroxynonénal, aldéhyde provenant du catabolisme des lipoperoxydes (Chen J et al., 2001).
A. Troubles ioniques
Ils sont dus à des dysfonctionnements des canaux transporteurs d’ions, tant dans la membrane cellulaire extérieure que dans les membranes des mitochondries.
1. La surcharge calcique mitochondriale
a. Changements ioniques dans le cytosol
Les lésions irréversibles du myocarde sont précédées d’une montée en Ca2+; les médicaments qui ralentissent cette montée ralentissent les processus nécrotiques ; d’abord contestée, cette observation a mis d’accord un grand nombre de chercheurs, ces dernières années (Murphy et Steenbergen, 2008).
b. Changements ioniques dans les mitochondries
La combinaison d’une élévation calcique avec une poussée de ROS peut déclencher l’ouverture du mégapore mPTP. Un phénomène se produisant dans la cellule menacée, la montée des ions Na+, peut avoir des conséquences positives sur la concentration de l’ion calcique et donc négatives sur la survie de la cellule, et l’application d’amiloride, un inhibiteur de ce phénomène, peut avoir des effets favorables sur l’évolution des effets ischémiques (Murphy E et al., 1991).
La captation du calcium cytosolique par les mitochondries se fait par l’intermédiaire du canal calcique uniporteur (figure III-1).
c. Cas des mitochondries myocardiques
Ces organelles se groupent en 2 populations (Chen Q et al., 2007) :
Les mitochondries SSM pâtissent plus rapidement de l’ischémie (Ueta et al., 1990; Lesnefsky et al., 1997). Selon Kim et al. (2006), c’est la formation de ROS qui provoquerait l’ouverture du mPTP, enclenchant les processus apoptotiques (voir première partie, chapitre VII), et qui serait alors suivie par les troubles calciques.
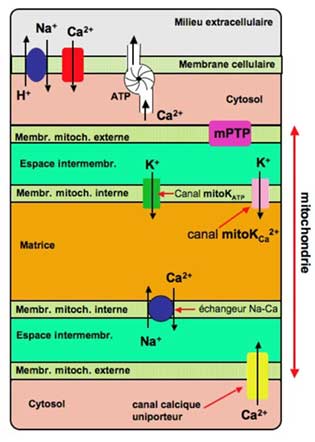
| Fig. III-1 : Coupe transversale schématisée de mitochondrie in situ montrant les canaux mis en jeu au cours des troubles de la reperfusion. En bleu, traversant la membrane cellulaire, l’échangeur sodium-proton (NHE : Na+-H+ Exchange). Dans la membrane mitochondriale interne, en bleu, l’échangeur sodium-calcium (NCX : Na+-Ca2+ Exchange). Pour plus de détails, voir partie 1, chapitre V. |
2. Rôle des canaux mitoK+ATP
Les canaux mitoK+ATP traversent la membrane mitochondriale interne. Ils permettent la sortie du potassium et leur ouverture est reconnue unanimement comme favorable à la récupération. Le potassium favorise le swelling des mitochondries. L’inhibition de la sortie de K+ par des inhibiteurs des canaux K+ATP bloquent la protection fournie par le préconditionnement : la glibenclamide et l’acide 5-hydroxydécanoïque suppriment ainsi les effets bénéfiques du préconditionnement (O’Rourke, 2004), tandis que le diazoxyde, qui promeut l’ouverture des canaux potassiques, induit un préconditionnement sans pré-ischémie. Des canaux semblables, obéissant aux mêmes agents pharmacodynamiques, traversent la membrane cellulaire et permettent la sortie du potassium cytosolique. On a montré que la bradykinine protège le cœur contre les lésions d’ischémie–reperfusion (Oldenburg et al., 2004) par l’ouverture de ces canaux, grâce à une chaîne de réactions biochimiques (Quinlan et al., 2008).
Ces canaux apparaissent de plus en plus comme des voies importantes pour lutter contre la nécrose myocardique et divers médicaments ont été proposés pour les ouvrir, tel le nicorandil, un médicament à double action ouvrant le canal mitoK+ATP et exerçant un effet vasodilatateur (donneur de •NO) (Miura et Miki, 2003). Voir chapitre IV.
3. Rôle des canaux mitoK+Ca2+
Ces canaux abaissent également la concentration intra-mitochondriale en potassium et leur activation permet d’obtenir un préconditionnement pharmacologique (Xu et al., 2002). On dispose d’un inhibiteur de ce canal, la paxilline.
B. L’ouverture du mPTP menant à l’apoptose
Voir partie I, Mitochondries, chapitre VII.
On considère actuellement que cette ouverture est le phénomène majeur durant l’ischémie-reperfusion (Ananthakrishnan et al., 2009). On a découvert qu’un des agents enzymatiques impliqués dans cette ouverture, durant l’ischémie-reperfusion cardiaque, était l’aldose réductase (Ananthakrishnan et al., 2009).
De nombreux travaux aux résultats contradictoires mettaient en doute l’effet létal des ROS parce que les « antioxydants » ne protégeaient pas efficacement (en admettant que ces merveilleux antidotes pénétrassent dans la cellule) ou encore parce que les ROS elles-mêmes n’étaient pas mises en évidence de manière concluante (on croyait dur comme fer que les techniques de détection à la TBA étaient valables ou que la mesure des lipoperoxydes par les diènes conjugués était pratiquée avec des instruments suffisamment sensibles, ce qui n’était pas encore le cas). Les recherches n’étaient donc pas concluantes. Un peu hâtivement, on déduisit de ces constatations que « la nécrose de reperfusion n’existait pas » (Piper et al., 1998).
Survinrent les travaux de l’équipe de Paradies et Petrosillo (Paradies et al., 1999 et 2000) qui démontrèrent que le primum movens de l’apoptose intrinsèque commence dans les mitochondries avec la libération du cytochrome c dans le cytosol, grâce à l’ouverture du mPTP. Cette sortie du cytochrome c nécessite la séparation de la protéine hémique d’avec une cardiolipine mitochondriale qui s’effectue par peroxydation des acides gras non saturés, processus initié par les ROS (Petrosillo et al., 2003b).
Aujourd’hui, la plupart des chercheurs reconnaissent que les ROS jouent un rôle initiateur favorable dans les processus de préconditionnement (Penna et al., 2009a). En effet, nous verrons dans le chapitre IV que l’ouverture (bénéfique) du canal K+ATP est due en partie à des ROS (comme le •NO) et que cette ouverture est inhibée par les antioxydants. Toutefois, il est avéré que certaines ROS, comme le peroxynitrite, sont toxiques et aggravent la situation.
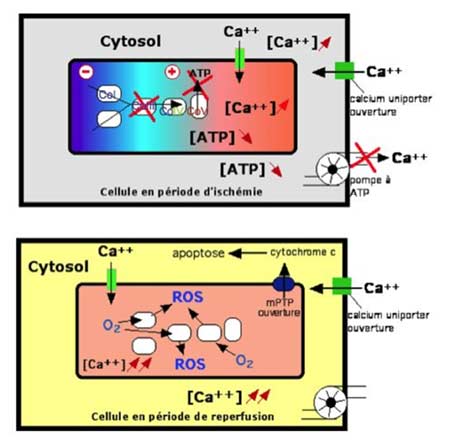
| Fig. III-2 : Phénomènes apparaissant dans les deux phases de l’ischémie-reperfusion. Au-dessus : suppression du transport d’électrons le long de la chaîne respiratoire (modification du potentiel membranaire ΔΨm), entraînant la suppression de la synthèse d’ATP qui aura comme conséquence la mise hors service des pompes refoulant le Ca2+ hors de la cellule, la montée de la concentration cytosolique en Ca2+ provoquant l’entrée du Ca2+ dans la mitochondrie. En dessous : l’entrée du Ca2+ continue, mais l’arrivée d’O2 au niveau des complexes I, III et IV provoque la synthèse de ROS en excès. Cet excès, combiné à une forte concentration en Ca2+, détermine l’ouverture du mPTP, d’où une kyrielle d’évènements létaux commençant par la sortie du cytochrome c et aboutissant à l’apoptose. |
La présence de ROS a été mise en évidence durant l’ischémie par RPE indirecte (spin traps) (Arroyo et al., 1987; Garlick et al., 1987; Bolli et al., 1988; Zweier, 1988) et par RPE directe (Zweier et al., 1987). Plus tard, d’autres travaux confirmèrent ces observations (Becker et al., 1999; Kevin et al., 2003; Becker, 2004).
La génération de ROS et de RNOS durant la période ischémique est faible, mais a été observée sur cardiomyocytes en culture et sur cœur isolé (Villa et al., 1994; Becker et al., 1999; Becker, 2004; Kevin et al., 2003).
Depuis quelques années, il ne fait plus aucun doute qu’une production de ROS anormalement élevée se produit au cours de la reperfusion (Arroyo et al., 1987; Garlick et al., 1987). Ce sont surtout des RNOS (dont le •NO) qui sont produits (Mason et al., 2000; Espey et al., 2002; Donzelli et al., 2006).
En 2004, Petrosillo et al. ont suggéré le mécanisme suivant pour décrire la cascade de phénomènes menant à l’apoptose cellulaire :
C’est manifestement pendant la réoxygénation qu’apparaissent abondamment les ROS : ce fut démontré non seulement au cours d’expérimentations animales (Bolli et al., 1989), mais aussi chez l’homme au cours de la thrombolyse (Beard et al., 1994), au cours d’angioplasties coronaires (Roberts et al., 1990) ou d’interventions à cœur ouvert (Kim et al., 1994).
Les ROS et les RNOS deviennent plus abondantes dès les premières minutes de la reperfusion (Wink et al., 1994; Wink et Mitchell, 1998; Wink et al., 1998; Wink et al., 1999; Grisham et al., 1999; Mason et al., 2000; Espey et al., 2002; Ridnour et al., 2004a et 2004b; Hare et Stamler, 2005; Donzelli et al., 2006).
3. Importance du peroxynitrite
L’existence du peroxynitrite (ONOO-) dans les tissus a acquis de plus en plus de vraisemblance. Depuis sa découverte en système biologique par Beckman et al. en 1990, la réalité de son existence dans les tissus a subi de nombreux avatars. Mais depuis la fin de cette décennie, les arguments en faveur de son existence et de sa haute toxicité pour les cellules se sont multipliés (Ronson et al., 1999; Ferdinandy et Schulz, 2003; Pacher et al., 2007). Dans deux revues parues en 2008 et 2009 (Penna et al., 2008 et 2009a), ONOO- est présenté comme un agent contribuant aux lésions de reperfusion.
D’autres chercheurs ont montré l’importance du phénomène inflammatoire durant la reperfusion : activation du complément provoquant la cytotaxie des leucocytes neutrophiles, avec déversement par ceux-ci de ROS et myéloperoxydase (Park et Lucchesi, 1999). Rappelons que ces cellules de défense agissent non seulement en déversant des protéases, mais aussi des ROS, et que la myéloperoxydase, abondante dans les neutrophiles, est une source de ROS particulièrement agressives.
Des lapins déficients génétiquement en protéines du complément montrent une nette réduction de la taille de l’infarctus consécutif à des manœuvres d’ischémie-reperfusion (Yasojima et al., 1998a et 1998b).
L’ischémie-reperfusion détermine l’adhésion des leucocytes neutrophiles
L’ischémie-reperfusion provoque l’adhésion des neutrophiles aux cellules endothéliales, augmentant le phénomène de « no-reflow » ; cette tendance à l’adhésion est potentialisée par le TNFα (Kokura et al., 2000) en provenance des lymphocytes T, durant les syndromes inflammatoires.
Ce fut d’abord au niveau hépatique que le rôle joué par les neutrophiles dans l’aggravation du syndrome d’ischémie-reperfusion fut mis en évidence (Jaeschke et al., 1990).
L’importance des leucocytes neutrophiles dans la taille de l’infarctus fut d’abord démontrée histologiquement, la gravité de l’infarctus étant proportionnelle à l’accumulation de ces cellules inflammatoires (Romson et al., 1983) ; comme on peut s’y attendre, la neutropénie et l’inhibition de l’adhésion des neutrophiles jouent également un rôle favorable (Simpson et al., 1988; Werns et Lucchesi, 1988).
Le jeûne aggrave le stress oxydant dans les mitochondries hépatiques au cours de l’ischémie-reperfusion (Domenicali et al., 2001).
Troubles de l'oxygénation et mitochondries - Chapitre IV: Ischémie-reperfusion : traitements |
Mitochondries et métabolisme de l'oxygène - Introduction |
Courrier |
 |
 |
 |