 Université de Liège - Centre de l'Oxygène, Recherche et Développement (CORD)
Université de Liège - Centre de l'Oxygène, Recherche et Développement (CORD) Université de Liège - Centre de l'Oxygène, Recherche et Développement (CORD)
L'Oxygène et la Vie: Tome II - L'Oxygène en Pathologie des Mammifères
Mitochondries et métabolisme de l'oxygène
Seconde partie : Troubles de l'oxygénation et mitochondries
Carol Deby
| Note: pour la facilité de la lecture, 1. chaque référence dans le texte comporte un lien vers les pages de bibliographie 2. les abréviations et les formules chimiques sont reprises dans les pages du glossaire ; elles sont également identifiées directement dans le texte (apparition en arrière plan lors du pointage de la souris) |
La mitochondrie : une cible incontournable de la cardioprotection du myocarde ischémique (Argaud et al., 2006).
La mitochondrie est à la fois une cible pour les ROS et une source de ceux-ci. Un concept émerge parmi d’autres depuis quelques années : une modulation du métabolisme oxydant mitochondrial, durant l’ischémie ou très précocement au cours du début de la reperfusion, pourrait protéger les fonctions mitochondriales et abaisser la mortalité des cellules myocardiques (Chen Q et al., 2007).
Pratiquée avant ou durant l’ischémie, cette modulation fait partie du préconditionnement ; intervenant dans les secondes qui suivent la reperfusion, elle constitue un postconditionnement.
Le préconditionnement peut être réalisé simplement par ischémie de durée suffisamment courte pour ne pas léser les cellules ou par agents pharmacodynamiques.
Nous avons vu précédemment les effets nocifs du rétablissement de l’arrivée d’oxygène dans un tissu trop longtemps ischémié. La reperfusion étend les lésions tissulaires : c’est la nécrose de reperfusion, concept bien admis en ce début de siècle (Argaud et al., 2006).
Au cours de nombreuses recherches, on a essayé de limiter les dégâts occasionnés par cette manoeuvre inévitable et mis au point des traitements permettant d’atténuer la sévérité de la phase de réoxygénation. Depuis une décennie, il semble de plus en plus certain que, par certaines manœuvres, on puisse réduire les dommages de la reperfusion.
Lorsque le traitement précède l’accident vasculaire (intervention sur les coronaires risquant de provoquer un arrêt de la circulation), il s’agit de préconditionnement, opération préventive destinée à provoquer une réaction dans les tissus qui leur permette d’affronter l’ischémie. Les manœuvres d’atténuation des lésions déjà survenues constituent le postconditionnement.
1. Définition
Dès 1986, Murry et al. ont montré expérimentalement que des ischémies brèves et répétitives (séparées de quelques minutes), précédant une ischémie de durée suffisante pour amener des phénomènes nécrotiques, réduisaient de manière significative l’étendue de l’infarctus. On désigne ces manœuvres sous le sigle IPC (ischemic preconditioning). Depuis, on réalise des ischémies cérébrales chez le Rat en clampant l’artère cérébrale médiane. Une occlusion de 10 minutes ne cause aucun dommage cérébral, mais induit une forte protection contre des ischémies-reperfusions ultérieures sévères (Currie et al., 2000). Des ischémies de préconditionnement sont à la base des travaux sur l’infarctus cardiaque, par clampage d’artère coronaire.
Cette thérapeutique préventive a été appliquée par après à d’autres animaux, dont l’homme, la souris, le lapin et le porc (Edwards et al., 2000; Kirino, 2002).
Penna et al. (2009a et 2009b) ont montré une curieuse évolution dans le temps de la résistance du myocarde à la reperfusion.
Première fenêtre (EPC : early preconditioning) : la protection commence immédiatement après la manœuvre de préconditionnement et dure de 2 à 3 heures. Suit une période de 12 à 24 heures durant laquelle la protection cesse.
Deuxième fenêtre (LPC : late preconditioning) : la protection réapparaît pendant une durée s’étendant de la 24ième heure à la 72ième heure après la manœuvre de préconditionnement.
Le préconditionnement peut être obtenu en pratiquant l’ischémie de courte durée sur un autre organe du même organisme.
3. Mécanismes
L’importance des mitochondries a été démontrée expérimentalement par le blocage de la chaîne de transport mitochondriale d’électrons (partie 1, chapitre III), peu avant de provoquer l’ischémie, ce qui réduit la respiration mitochondriale, augmente le transport des ions K+, mais accroît la production d’H2O2 (da Silva et al., 2003). Cette manœuvre atténue significativement l’étendue des dommages dans les tissus irrigués (Chen Q et al., 2007). Les mécanismes mis en jeu dans cette opération visent avant tout à empêcher l’ouverture du mégapore, le mPTP, qui débouche sur l’apoptose, et concernent au moins quatre agents:
a. Les canaux potassiques ATP-dépendants de la membrane mitochondriale interne, K+ATP (Zhao et al., 2009)
b. Les canaux potassiques Ca2+-dépendants de la membrane mitochondriale interne, K+Ca2+
c. Certaines ROS comme le monoxyde d'azote, •NO
d. Les « heat shock proteins » (HSP) (pour un article général sur les HSP, voir Wirth et al., 2002)
Dhodda et al., en 2004, se livrèrent à une étude génomique et protéomique pour comprendre le mécanisme de cette protection et mirent en évidence une expression accrue de certaines « heat-shock-proteins », dont HSP70, HSP27 et HSP90, de la guanylylcyclase (Friebe et al., 2003), de la muskeline, du récepteur au facteur d’activation plaquettaire et de la β-actine. L’efficacité du clampage de préconditionnement s’accroissait en fonction du temps séparant le préconditionnement de l’ischémie de longue durée. : les rats traités avaient une réduction de 38% de la taille de la zone nécrotique par rapport aux témoins clampés, mais non conditionnés, si le clampage de 10 minutes était pratiqué 3 jours avant la réalisation de l’ischémie-reperfusion.
e. la redistribution de l’hexokinase depuis le cytosol vers les mitochondries lorsque les cellules se trouvent en conditions hypoxiques ; une des premières tentatives d’autoprotection est de protéger la glycolyse (Murry et al., 1986). C’est une des réactions de prévention de l’apoptose (Zuurbier et al., 2005).
Il semble cependant que ce n’est pas le blocage de l’ETC qui assume l’effet protecteur (Tanaka-Esposito et al., 2007), bien que Khaliulin et al. (2004) aient attribué le succès du préconditionnement à la diminution des oxydations dans les mitochondries. Plus récemment toutefois, le blocage réversible de l’ETC a été tenu pour responsable de l’effet bénéfique du préconditionnement (Chen Q et al., 2006).
Pour Glanemann et al. (2003), le préconditionnement ischémique préserverait la microcirculation et stabiliserait l’état redox, tout au moins au niveau du foie. Tout récemment, le rôle bénéfique de l’amobarbital, qui intervient dans l’homéostasie de l’état redox et réduit la surcharge matricielle en Ca2+, a été confirmé (Aldakkak et al., 2008).
4. Importance de la température
En 2007, Khaliulin et al. ont insisté sur le rôle de la température comme agent préconditionnant, agissant vraisemblablement par action sur l’ouverture du mégapore.
5. Importance de la pression d’oxygène durant la reperfusion
Le recours à un abaissement de la pO2 au cours de la reperfusion (150 mm Hg pendant 3 minutes, puis remontée à 600 mm Hg) permet de protéger les cardiolipines de la peroxydation (Petrosillo et al., 2005).
1. Quelques types de drogues préconditionnantes
Elles déclenchent toutes des signaux aboutissant à l’ouverture du canal potassique ATP-dépendant, K+ATP
Outre l’ischémie préparatrice, divers travaux ont préconisé l’usage de cardioprotecteurs dès le début de la reperfusion. On dispose là d’une fenêtre d’intervention très étroite (Yang et al., 2004 ; Tang et al., 2006 ; Murphy et Steenbergen, 2008). L’ultime action de ces substances pharmacodynamiques est de stimuler les agents énumérés au paragraphe précédent.
Les premières recherches ont utilisé l’adénosine, les opioïdes et la bradykinine. Dès 1984, l’attention des cliniciens était attirée sur le rôle protecteur assumé par certains opioïdes, tels que la morphine, le fentanyl et certains de ses dérivés (alfentanyl, sulfentanyl) (Bovill et al, 1984). Une étude de Thomson et al., en 2000, concluaient que dans les bypass des artères coronaires et les bypass cardio-pulmonaires (CPB), le fentanyl et le sufentanil avaient des effets favorables sur la récupération des malades. En 2007, Murphy et Steenbergen montraient la supériorité de la morphine sur le fentanyl en pré-CPB sur la récupération des fonctions ventriculaires après intervention. Une explication de l’effet des opioïdes fut fournie par une potentiation de l’ouverture du canal K+ATP. Les anesthésiques volatils (isoflurane) exercent la même action et montrent un effet agoniste (Patel et al., 2002). Propofol et midazolam se sont avérés nettement inférieurs aux fluranes du point de vue récupération de la fonction ventriculaire et diminution de la sortie de troponine I (marqueur de lésions myocardiques) (De Hert et al. 2004).
Tous ces agents nécessitent des récepteurs qui comprennent une partie extracellulaire à laquelle ils se lient en déclenchant le signal qui traverse la membrane le long de la protéine et aboutit à une protéine spéciale pouvant servir d’interrupteur, la protéine G (figure IV-1). De là, le signal est orienté selon diverses voies (Downey et al., 2005 et 2007). Il est de plus en plus admis que les canaux K+ATP jouent un rôle primordial au cours du préconditionnement, par diminution du « swelling » de l’espace intermembranaire ; leur ouverture permettrait l’évacuation de l’excès de Ca2+ et préviendrait ainsi l’ouverture du mPTP (Miyamae et al., 1996 ; Tamura et al., 2005).
Les signaux en provenance des récepteurs sont orientés vers une enzyme phosphorylante, la phosphokinase Cε (PKCε) laquelle phosphoryle les canaux K+ATP mitochondriaux et cellulaires (Weber et Schlack, 2008). On sait que la PKCε peut activer la •NO synthase (NOS); or les inhibiteurs de la NOS diminuent le préconditionnement par opioïdes, de même que les anti-ROS (génistéine, propofol, etc…) ; en considérant que l’ouverture du canal K+ATP mitochondrial est activée par les ROS et le •NO, on peut admettre que la PKC agit directement sur le canal potassique et indirectement par activation des •NO synthases produisant du •NO (Weber et Schlack, 2008). La redistribution de l’hexokinase vers les mitochondries est produite par les opioïdes (Zuurbier et al., 2005).
La PI3 kinase active également diverses kinases ; la fin de la cascade est l’activation de l’Akt, facteur essentiel pour la production de ROS et RNOS.
La voie classique de signalisation est celle passant par les protéines PI3K-Akt ; elle fut proposée par Tong et al. en 2000. Le stimulus IPC provoque la phoshorylation de l’Akt et de sa cible GSK3, étapes biochimiques essentielles pour l’IPC (Tong et al., 2002).
Les phosphorylations sont des réactions enzymatiques dominantes dans la transmission des signaux. Un groupe phosphate -PO3= est transféré d'une molécule à une autre, porteuse d'un groupe polaire (aminoacide par exemple). Souvent, la molécule donneuse de phosphate est l'ATP.
Les phosphorylations sont catalysées par les protéines kinases PI3kinases, les Akt et notamment le pAkt.
Ce serait via l’activation des récepteurs à l’adénosine A3 qu’agirait le préconditionnement par le resvératrol (Das et al., 2005).
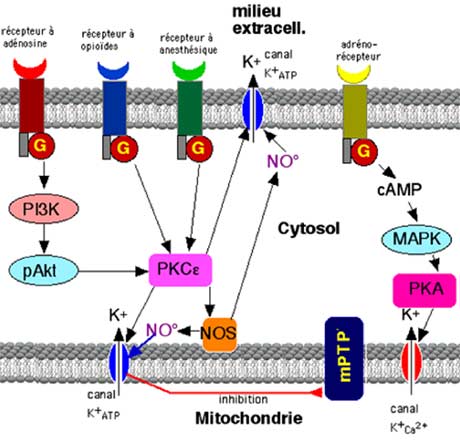
| Fig. IV-1 : Les principales voies d’activation du canal K+ATP menant à la neutralisation du mPTP, inhibant ainsi l’apoptose. Certaines étapes ne figurent pas sur ce schéma très simplifié. Le lecteur est invité, pour plus de détails, à lire Murphy et Steenbergen (2008), ainsi que Lochner et al. (2009). G : protéine G ; cAMP : adénosine monophosphate cyclique ; PKC : protéine kinase C ; PKA : protéine kinase A ; PI3K : phosphatidylinositol-3 kinase ; MAPK : mitogen-activated protein kinase. |
d. Activation des adrénorécepteurs
Rôle de la voie α-adrénergique:
Le signal a-adrénergique aboutit à la PKC (figure IV-1). Les chercheurs utilisèrent soit un inhibiteur de la voie α-adrénergique (prazosine), soit des inhibiteurs spécifiques de la PKC (chelérythrine et bisindolylmaléimide), ou des médicaments imitant la stimulation α-adrénergique (phényléphrine). Aucun résultat ne fut obtenu provoquant une modification des effets du préconditionnement.
Rôle de la voie β-adrénergique:
Par contre, la montée de l’AMP cyclique (cAMP) était en faveur d’une action de la voie β-adrénergique : effectivement, l’usage d’agonistes et d’antagonistes de cette voie démontra que :
Diverses équipes ont étudié le préconditionnement β-adrénergique, disséqué par Lochner et al. (Asimakis et al.,1994; Nasa et al., 1997; Robinet et al., 2005).
Depuis quelques années, on s’est aperçu que, durant la reperfusion, des kinases de signalisation étaient activées, que cette activation était amplifiée par le préconditionnement et qu’elle exerçait une action cardioprotectrice (Hausenloy et al., 2005a et 2005b; Hausenloy et Yellon 2006).
3. Rôle du calcium dans le préconditionnement
Rappelons que les voies d’entrée du calcium dans les mitochondries sont principalement le canal d’échange Na+/H+ (NHE : Na+/H+ exchanger) et le canal calcique unipolaire. La figure IV-2 rappelle l’importance du Ca2+, des ROS et du mPTP dans la mort cellulaire.
Talukder et al., en 2009, ont utilisé un inhibiteur du NHE, le cariporide SEA0400 (Namekata et al., 2006). Le traitement par cet inhibiteur apporte une certaine protection.
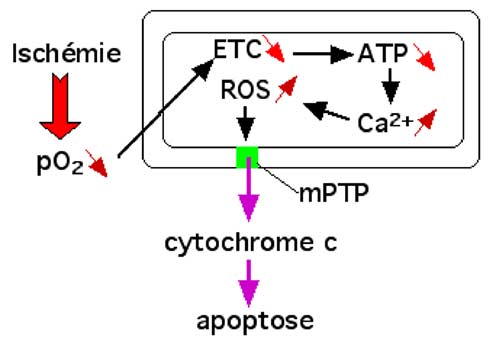
| Fig. IV-2 : L’ischémie entraîne une chute de la pO2 dans les cellules et leurs organelles. Dans les mitochondries, la chaîne de transport d’électrons (ETC) se paralyse tandis que la production d’ATP s’effondre, au détriment de la pompe à calcium qui cesse de refouler ces ions hors de la matrice. La concentration en ions calciques s’élève, favorisant la montée des ROS. Celles-ci ouvrent le pore mPTP et permettent la sortie du cytochrome c, libéré de ses attaches avec les cardiolipines, peroxydées par les ROS. L’apoptose est amorcée. |
4. Effets d’enzymes anti-ROS
L’administration d’enzymes anti-ROS (catalase, superoxyde dismutase), 15 minutes avant l’ischémie améliore la situation du myocarde lors de la reperfusion ; ces enzymes sont sans effet lorsqu’elles sont appliquées après la reperfusion (Park et Lucchesi, 1999). Ces faits apportent une nouvelle confirmation que c’est pendant la réoxygénation que se joue le drame. Toutefois, l’effet protecteur de la catalase et de la SOD avait été contesté (Gallagher et al., 1986 ; Uraizee et al., 1987), mais pour certains cette association serait efficacement protectrice (Naslund et al., 1986).
Plusieurs auteurs préconisent les superoxyde dismutases pour réduire l’ampleur des dommages de l’ischémie-reperfusion (Chi et al., 1989 ; Jin et al., 2005) ; des SOD de synthèse (SOD mimetics) ont été présentées comme cardioprotectrices (Kilgore et al., 1994).
C’est probablement par une action anti-ROS catalytique que l’on explique le rôle protecteur joué par le resvératrol administré préventivement vis-à-vis de l’ischémie cérébrale (Della-Morte et al., 2009).
5. Rôle de l’héparine
Administrée 2 heures avant l’ischémie, la N-acétyl-héparine réduit de 43% les dimensions de l’infarctus (Friedrichs et al., 1994); non seulement l’héparine est anticoagulante, mais elle inhibe la cascade du complément (Cruse et Lewis, 1993).
En pratique, l’intervention thérapeutique est le plus souvent appliquée après un accident cardiaque ou cérébral, donc bien après l’installation des troubles ischémiques. Le post-conditionnement, s’il est réellement possible, reposera sur d’autres considérations. Le « postconditioning » intervient seulement durant les premières minutes de la reperfusion.
Diverses considérations ont été récemment avancées : généralités sur le postconditionnement du cœur (Staat et al., 2005), comparaison des résultats obtenus avec le pré- et le postconditionnement (Zhao et al., 2003) ; une très bonne revue des travaux en cours sur ce sujet est parue, attribuant le rôle principal du postconditionnement à l’empêchement de l’ouverture du mPTP (Gateau-Roesch et al., 2006).
Récemment une stratégie « mécanique » consistant en de courtes reperfusions, interrompues par de courtes périodes d’ischémie (ou d’hypoxie), a été proposée (Vinten-Johansen, 2007).
En cas d’accident vasculaire, la tentative de reperfusion (thrombolyse, angioplastie, chirurgie cardiaque) ne commence généralement qu’après des heures d’anoxie tissulaire. La protection ne survient qu’après l’accident et vise à limiter les lésions de reperfusion. Ce phénomène est nommé par Braunwald et Kloner (1985) « lethal reperfusion-induced injury ».
Hausenloy et al. se sont penchés sur une voie de signalisation qu’ils estiment bénéfique : celle des kinases (Hausenloy et al., 2005a et 2005b) ; ces chercheurs ont inventé une nouvelle expression: « reperfusion injury salvage kinase » (RISK) (Hausenloy et Yellon, 2004).
Cette voie comprend les étapes suivantes : la PI3K (phosphatidylinositol-3-OH kinase)-Akt et les p42/p44 extracellular signal-regulated kinases (Erk1 & 2).
Les protéines de la voie RISK protègeraient les tissus durant la reperfusion ; elles exercent une activité antiapoptotique (Hausenloy et al., 2005a et 2005b).
Inhibition de la p38MAPKinase
L’ischémie-reperfusion provoque la phosphorylation du VDAC par la p38MAPK, résultant en une dérégulation du trafic ionique entre le cytosol et l’espace intermembranaire, et une menace d’apoptose ou de nécrose (Schwertz et al., 2007). Cependant, les recherches de Moolman et al. (2006) ont donné des résultats plus nuancés : la p38MAPK ne jouerait un rôle nocif que dans les cas du préconditionnement par ischémie et n’interviendrait pas dans le préconditionnement pharmacologique.
L’ouverture du mPTP favorise la nécrose ou l’apoptose de la cellule; elle survient durant les premières minutes de la reperfusion (Lim, 2007). Les manœuvres de pré- et post-conditionnement aboutissent toutes à ce but final : inhibition d’ouverture du mégapore. Plusieurs travaux ont mis en évidence l’utilité de la cyclosporine, inhibiteur de l’ouverture du mPTP, et d’autres agents empêchant la transition de perméabilité mitochondriale (Nazareth et al., 1991; Schreiber et Crabtree, 1992; Kerr et al., 1999; Argaud et al., 2004; Piot et al., 2008; Javadov et al., 2008) .
Cela a été vérifié au cours de la cardioprotection produite par les manœuvres de pré-et postconditionnement qui empêchaient cette ouverture (Javadov et al., 2003; Hausenloy et al., 2004 a et 2004b; Juhashova et al., 2004; Argaud et al., 2005a et 2005b; Bopassa et al., 2006).
Une voie originale de recherche a été ouverte en cancérologie, en recherchant des agents pouvant provoquer l’ouverture des mPTP dans les cellules cancéreuses, entraînant ainsi leur mort par apoptose (voir une revue sur la question dans Armstrong, 2006).
De nombreux autres organes ont été étudiés, le foie, le système nerveux central, le poumon et les muscles, avec la même conclusion : c’est le mégapore qui doit être la cible des thérapeutiques de préservation.
◊ Essais d’inhibition directe du mégapore
La cyclosporine A et la sanglifehrine A s’attaquent à la cyclophiline qui commande le mPTP. Chez les souris exposées à des manœuvres d’ischémie-reperfusion et traitées par ces substances, on observe une réduction de la taille des zones infarcies, tant dans le cerveau que dans le cœur (Schinzel et al., 2005; Leung et al., 2008).
◊ Action sur d’autres canaux échangeurs d’ions
On connaît le rôle favorable de l’inhibition du canal échangeur N+/H+ par l’amiloride (Karmazyn, 1988 et 1993) qui a été confirmé en 2001 (An et al., 2001).
4. Lutte contre les ROS
Des travaux contradictoires sont parus, beaucoup mettant en garde contre les espèces oxygénées activées, d’autres les présentant comme nécessaires. Plus paradoxale encore est cette publication vantant les effets protecteurs d’H2O2 dans l’ischémie-reperfusion cardiaque (Yaguchi et al., 2003).
Troubles de l'oxygénation et mitochondries - Chapitre V: Synopsis du rôle des ROS dans les affections cardiaques |
Mitochondries et métabolisme de l'oxygène - Introduction |
 |
 |
 |